Introduction
Dysferlin-deficient limb-girdle muscular dystrophy
type 2B (LGMD2B), distal Miyoshi myopathy (MM) and other less
frequent phenotypes constitute a group of recessive disorders known
as dysferlinopathies. MM with autosomal recessive inheritance and
localization to chromosome 2p is caused by autosomal recessive
mutations in the human dysferlin gene (DYSF) (1,2). In
cases of MM, the involvement of the gastrocnemius and soleus
muscles, which causes a difficulty in standing on one's tiptoes, is
characteristic. Clinical signs usually present for individuals
between the late teens and prior to the age of 30 years. According
to its clinical and histopathological manifestations, MM has been
categorized as a distal myopathy with typical symptoms of muscular
dystrophy (3,4). Distal MM is also known as Miyoshi-type
distal muscular dystrophy, and was first reported in 1986 by
Miyoshi et al (5).
Subsequently, an increasing number of cases of MM have been
reported, particularly in Japan, Brazil, Finland and South Korea
(6–8). In China, there has only been one report
of MM in a family in 2004 (9).
According to conservative estimates, the incidence of MM is
~l/440,000 in Japan (10). Among the
Jewish population in Libya, 10% have been found to be carriers of
the DYSF gene mutation (1,624 delG), and the incidence of
LGMD2B in adults from this population has reached l/1,300 (11,12).
However, the corresponding statistics in China remain unknown.
The present case report describes an atypical MM
patient, who was initially misdiagnosed with inflammatory myopathy
and finally was diagnosed with MM according to pathology,
immunohistochemistry and electromyography.
Case report
Patient admission and symptoms
This case report was approved by the ethical review
committee of the the Affiliated Hospital of Binzhou Medical
University (Binzhou, China), and the patient provided informed
consent. The patient, a 37-year-old man, was admitted to the
Affiliated Hospital of Binzhou Medical University in June 2013
presenting with slowly progressive weakness in the left foot, which
was first experienced while driving 1 month prior to admission.
Gradually, the patient developed difficulties in standing and
walking upstairs when applying pressure on his left foot, which
caused him to visit a doctor. The patient had a history of
hypertension. There was no relevant family genetic history.
Neurological examination
Neurological examination of the patient revealed
normal cranial nerve function and intellectual status. The patient
did not exhibit tremor or involuntary movements. Muscle tone,
muscle strength and sensation were normal. The tendon reflexes were
present and symmetrical. No ataxia or abnormal gait was
observed.
Laboratory examination
Laboratory examination results showed elevated blood
creatine kinase (CK) levels at 4,974.9 U/l (normal range, 25–200
U/l). Electromyography (EMG) was performed to investigate the
origin of the patient's muscle damage. EMG revealed myopathic motor
units and recruitment patterns. Initially, the patient's condition
was considered to be an inflammatory myopathy and he was treated
with dexamethasone (10 mg for 5 days followed by gradual tapering).
Following treatment, the CK levels temporarily dropped to 1,493.8
U/l; however, CK levels increased again following the reduction of
hormones, reaching 2,698.2 U/l, which suggested that there had been
no actual improvement. Pathological examination of the muscle was
then performed.
Pathological examination of the
muscle
Βiopsy tissue obtained from the left quadriceps
muscle was precooled in isopentane, frozen in liquid nitrogen, and
frozen muscle cross-sections of 8-mm thickness were thereby
produced. Routine histological and enzyme activity analyses of the
sections were conducted. Immunohistochemical and other types of
staining included: Hematoxylin and eosin (H&E), modified Gomori
trichrome (MGT), succinate dehydrogenase (SDH), cytochrome oxidase
(COX), Oil Red O (ORO), periodic acid-Schiff staining (PAS),
adenosine triphosphatase (ATPase), nicotinamide adenine
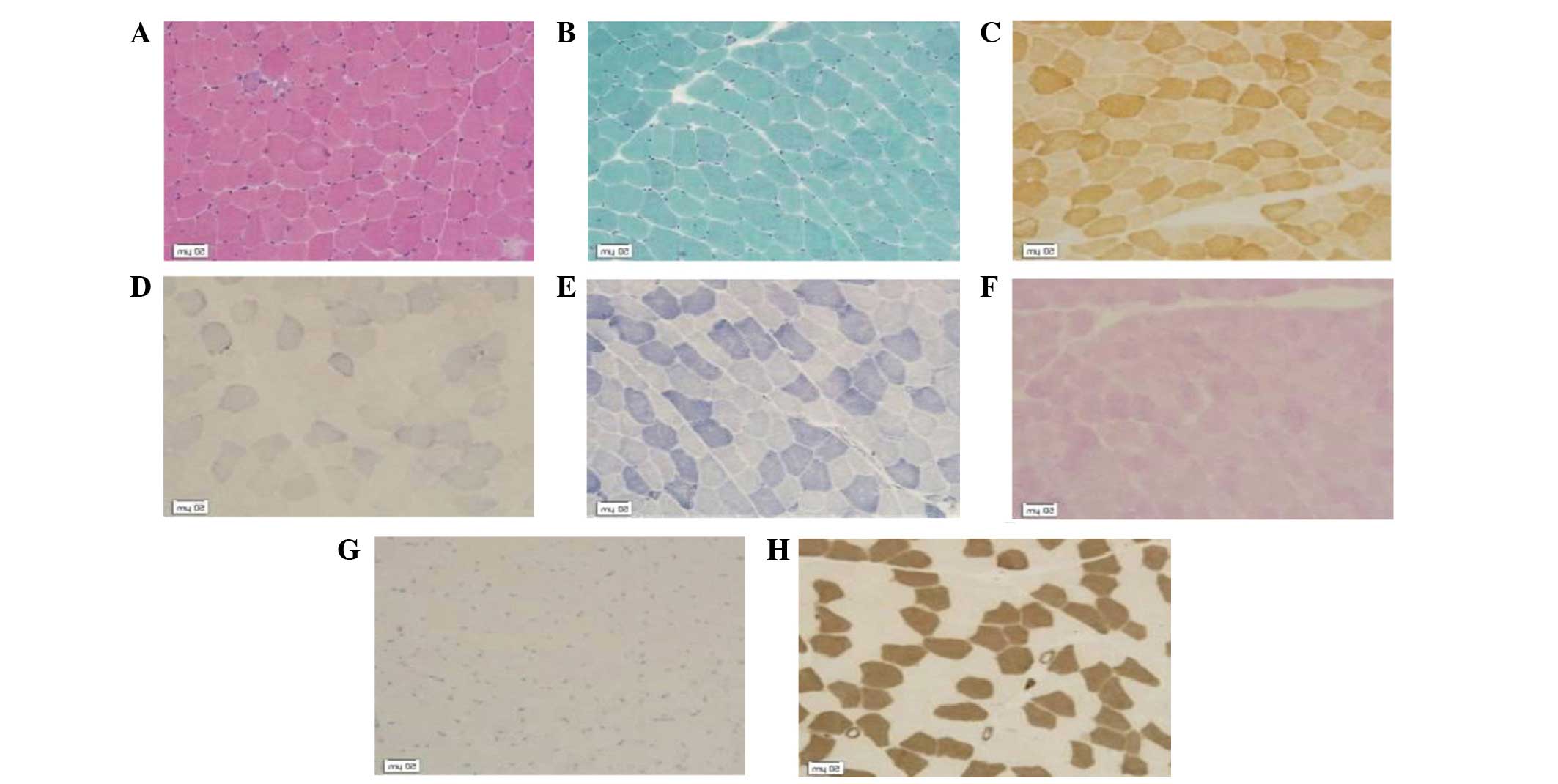
dinucleotide-tetrazolium reductase (NADH-TR; Fig. 1), dysferlin and dystrophin-C, –N and
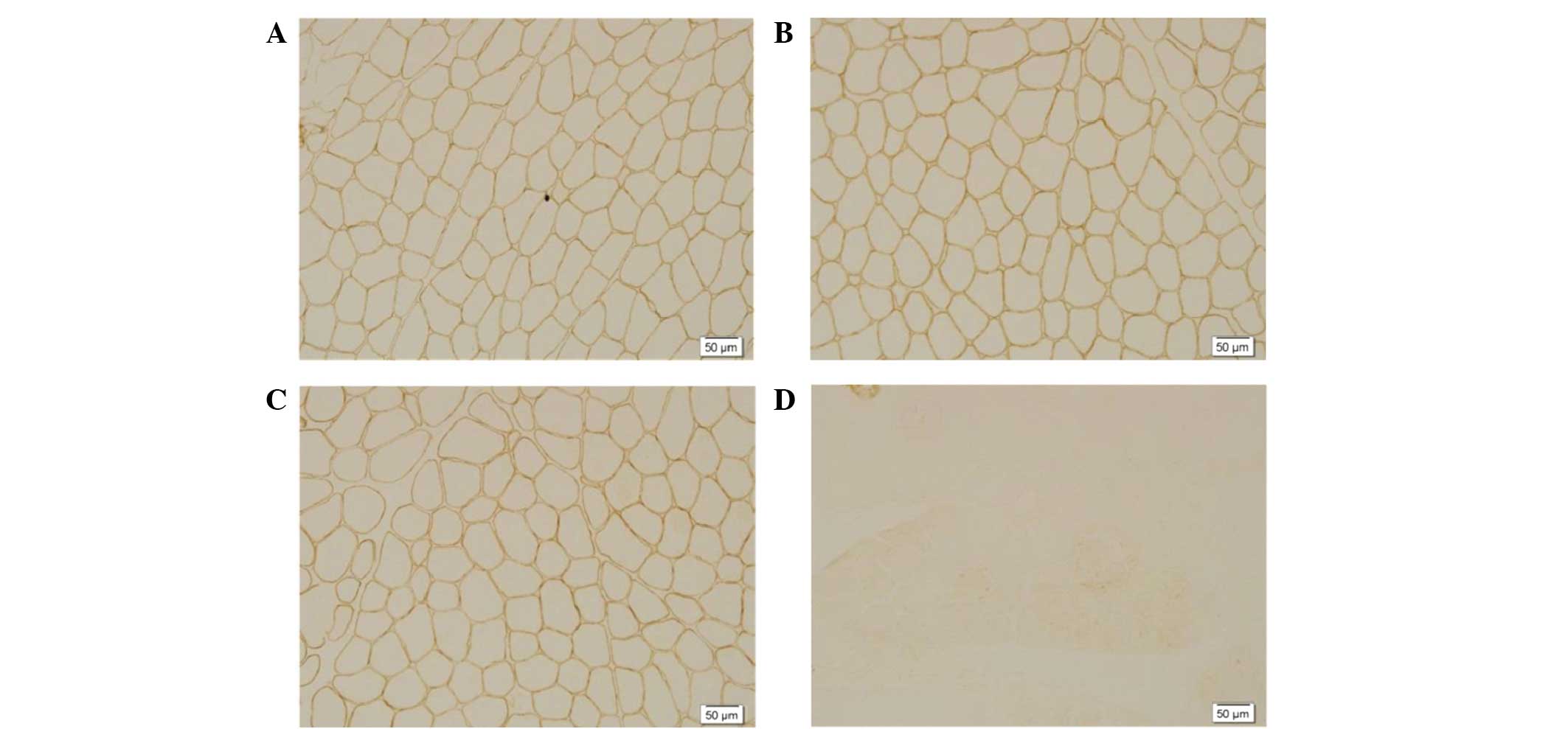
–R staining (Fig. 2).
The muscle fiber size was observed to vary markedly
compared with that of healthy muscle tissues. Necrotic and
regenerating muscle fibers were observed. No endomysium hyperplasia
or inflammatory cell infiltration was observed (H&E; Fig. 1A). Ragged red muscle fibres or rimmed
vacuoles were not shown by the MGT staining (Fig. 1B). Fiber enzyme activity was not
reduced (COX; Fig. 1C). Ribosome
biogenesis factors and spindle-shaped viruses were not detected
(SDH; Fig. 1D). Mesh structure
between myofibrils was found to be normal (NADH; Fig. 1E). The glycogen content of muscle
fibers was not increased (PAS; Fig.
1F). No lipid droplets were observed within the muscle fibers
(ORO; Fig. 1G). Type-I fibers were
distributed alternately with type-II fibers. (ATPase; Fig. 1H).
Immunohistochemical staining revealed positive
staining for dystrophin-C (Fig. 2A),
dystrophin-N (Fig. 2B) and
dystrophin-R (Fig.2C) and the total
absence of dysferlin labeling along the membrane (Fig. 2D).
In the present study, the pathology of
dysferlinopathy was characterized by changes of muscular dystrophy.
The detection of dysferlin deficiency or marked reduction on the
sarcolemma, as indicated by immunohistochemical staining using
anti-dysferlin monoclonal antibody, may thus be used in the
diagnosis of dysferlinopathy.
Discussion
Distal myopathy is a disease characterized by distal
limb muscle weakness. According to the onset age, affected muscles
and genetic factors, distal myopathy is divided into several
subtypes (13).
Congenital defect of dysferlin expression on muscle
cell membrane has been suggested to be associated with MM (4), which features a marked increase in
blood CK levels, compared with that in other subtypes of distal
myopathy. The significant dysferlin protein deficiency of the
present case, indicated by immunohistochemical staining of the
muscle, suggested distal myopathy. The differential diagnosis
criteria of common types of distal myopathy are shown in Table I.
 | Table I.Differential diagnosis of common types
of distal myopathy. |
Table I.
Differential diagnosis of common types
of distal myopathy.
| Variables | Nonaka myopathy | Miyoshi myopathy | Welander
myopathy | Finnish myopathy | Ophthalmo-pharyngeal
distal myopathy |
|---|
| Inheritance type | AR | AR | AD | AD | AR or AD |
| Onset age
(years) | 20–30 | 15–30 | >40 | >35 | <40 |
| Main muscles
involved | Anterior leg
muscles | Posterior calf
muscle | Upper limb | Anterior limb
muscles | Extraocular and
bulbar muscles, distal lower extremities |
| Creatine kinase | <5-fold | 10–100-fold | <5-fold | <5-fold | <5-fold |
| Rimmed vacuoles | (+) | (−) | (+) | (+) | (+) |
| Dysferlin
staining | (+) | (−) | (+) | (+) | (+) |
The patient of the present study reported no family
history of Miyoshi myopathy. One clinical manifestation in the
present case was the inability to stand on tiptoe on the left foot.
Progressively, the CK levels in the patient's peripheral blood
increased markedly. According to the classification of distal
myopathy presented in Table I, these
clinical manifestations are suggestive of a diagnosis of distal
MM.
MM is an autosomal recessive disease caused by a
DYSF gene mutation, located on chromosome 2p13 (1). Dysferlin protein is encoded by
DYSF. The DYSF gene mutation leads to a deficiency of
dysferlin proteins, which results in dysferlinopathy. The majority
of MM patients present with the initial symptoms between 15 and 30
years of age; but the patients have a broader range of ages of
onset (12–36 years) (14). In distal
MM, the initial symptoms are in the posterior compartment of the
distal lower extremity, which causes an inability to walk on
tiptoes or up stairs (15). Pain and
discomfort in the calves may also occur (16). The gastrocnemius muscles become
atrophic and the stretch reflexes of the ankle muscle are lost
(17). The tendency for early
involvement of the gastrocnemius muscles has been considered the
clinical hallmark of distal MM, which distinguishes it from other
distal dystrophies (13). However,
muscles of the anterior compartment of the distal lower extremities
eventually weaken as well (18). The
involvement of upper extremities is unusual in the early stages of
the disease (16). With the
progression of the disease, patients may develop proximal leg and
arm weakness to various degrees (16). The hamstring muscle group (knee
flexors) may become weaker than the quadriceps muscles (knee
extensors), which has implications for the choice of biopsy site
(19). Disease progression varies
from patient to patient, with some remaining moderately stable with
distal weakness, and others presenting a more aggressive pattern,
involving both proximal and distal muscles (5). A characteristic laboratory finding of
the disease is a marked increase in serum CK levels, up to
20–150-fold above normal (20). In
certain cases, an extremely high CK level may be detected during
routine blood tests before any clinical weakness or atrophy is
present (21,22).
Although dysferlinopathy is caused by a single
DYSF gene, it is well-known that dysferlinopathy has various
clinical presentations, such as distal MM, LGMD2B and distal
anterior compartment myopathy (Table
II). The early involved muscles determine the clinical
phenotype of dysferlinopathy. In the present case, the
gastrocnemius was the initially involved muscle, which matched the
symptoms of MM. In addition, the immunohistochemical analyses,
using an anti-dysferlin monoclonal antibody, showed a deficiency or
marked reduction in dysferlin on the sarcolemma.
| Table II.Clinical phenotypes of
dysferlinopathy. |
Table II.
Clinical phenotypes of
dysferlinopathy.
| Phenotype | Miyoshi myopathy | Limb-girdle muscular
dystrophy type 2B | Distal anterior
compartment myopathy |
|---|
| Major-muscle
involvement | Posterior calf
muscle | Proximal limbs and
calf muscle | Anterior leg
muscles |
| Muscle pathology |
|
|
|
| In
common | Muscle fiber
size | Muscle fiber
size | Muscle fiber
size |
|
Differences | Nuclear shift | Necrotic and
regenerating muscle fibers | Nuclear shift |
| Mutated gene | DYSF gene,
located in 2p13 | DYSF gene,
located in 2p13 | DYSF gene,
located in 2p13 |
| Responsible
protein | Dysferlin protein,
located in the muscle fiber membrane, to repair muscle fiber | Dysferlin protein,
located in the muscle fiber membrane, to repair muscle fiber | Dysferlin protein,
located in the muscle fiber membrane, to repair muscle fiber |
| Creatine kinase | 30–100-fold | 30–100-fold | 30–100-fold |
| Muscle immuno | Loss of
dysferlin | Loss of
dysferlin | Loss of
dysferlin |
| –histochemical
staining | protein staining | protein staining | protein staining |
In the present case, a 37-year old male patient
presented with the inability to stand on his left foot for 1 month
prior to admission to the hospital. Initially, the patient was
considered to have inflammatory myopathy, on the basis of an EMG
examination. Following treatment with hormone therapy, the CK level
temporarily dropped to 1,493.8 U/l, but subsequently increased to
2,698.2 U/l following the reduction in hormone administration, with
no obvious improvement of the symptoms. Subsequently, pathological
muscle examinations were conducted. The results of pathological
muscle examination, such as the muscle fiber size, and necrotic and
regenerating muscle fibers, as well as total absence of dysferlin
on the sarcolemma in immunohistochemical staining of muscle, played
an important role in diagnosing the patient with
dysferlinopathy.
In atypical MM, the following characteristics of the
present patient could lead to misdiagnosis: i) The patient was 37
years old, while MM is predominantly observed at a younger age
(15–20 years); ii) the onset time was short; 1 month in the present
case, in the report by Park et al (14) about typical MM, the course was 2–30
years; iii) the muscle involved was unilateral in the present
patient, but in the reports by Wang et al (16), Zhang et al (17) and Gayathri et al (23) typical MM was bilateral; and iv) the
present case was sporadic; however, typically MM is a rare
autosomal recessive inherited myopathy and sporadic cases are
uncommon.
Due to certain pathological features that are
observed in cases of MM, such as muscle weakness, elevation of CK
levels and mononuclear cell infiltration, it is not rare for the
disease to be misdiagnosed as inflammatory myopathy. A previous
study found that certain patients with MM also present with
gastrocnemius muscle myalgia, swelling and even false hypertrophy
at the early stage of the disease (12). The pathogenesis of these symptoms is
not clear and may be associated with transient inflammatory
reaction. The early symptoms of MM are not typical and are readily
misdiagnosed; therefore, the pathological muscle examination should
be undertaken as soon as possible. In particular,
immunohistochemical staining including anti-dysferlin monoclonal
antibodies may be useful for the identification of possible
dysferlin deficiency.
Acknowledgements
This study was supported by a grant from the Science
and Technology Planning Program of Binzhou Medical University of
China (grant. no. BY2012KJ03) and the Science and Technology
Development Planning Program of Binzhou City (grant. no.
2014ZC0144).
References
|
1
|
Weiler T, Greenberg CR, Nylen E, Halliday
W, Morgan K, Eggertson D and Wrogemann K: Limb-girdle muscular
dystrophy and Miyoshi myopathy in an aboriginal Canadian kindred
map to LGMD2B and segregate with the same haplotype. Am J Hum
Genet. 59:872–878. 1996.PubMed/NCBI
|
|
2
|
Liu J, Aoki M, Illa I, Wu C, Fardeau M,
Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, et al:
Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi
myopathy and limb girdle muscular dystrophy. Nat Genet. 20:31–36.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nonaka I: Distal myopathies. Curr Opin
Neurol. 12:493–499. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mastaglia FL, Lamont PJ and Laing NG:
Distal myopathies. Curr Opin Neurol. 18:504–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miyoshi K, Kawai H, Iwasa M, Kusaka K and
Nishino H: Autosomal recessive distal muscular dystrophy as a new
type of progressive muscular dystrophy. Seventeen cases in eight
families including an autopsied case. Brain. 109:31–54. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tagawa K, Ogawa M, Kawabe K, Yamanaka G,
Matsumura T, Goto K, Nonaka I, Nishino I and Hayashi YK: Protein
and gene analyses of dysferlinopathy in a large group of Japanese
muscular dystrophy patients. J Neurol Sci. 211:23–28. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oh SH, Kang SW, Lee JG, Na SJ, Kim TS and
Choi YC: Clinical and pathological characteristics of four Korean
patients with limb-girdle muscular dystrophy type 2B. J Korean Med
Sci. 19:447–452. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vainzof M, Anderson LV, McNally EM, Davis
DB, Faulkner G, Valle G, Moreira ES, Pavanello RC, Passos-Bueno MR
and Zatz M: Dysferlin protein analysis in limb-girdle muscular
dystrophies. J Mol Neurosci. 17:71–80. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun SC, Fan ZS, Wu HC, Leturcq F, Zhang
BF, Yu W, Deburgrave N, Liu M and Song YJ: Dysferlin deficiency:
The cause of limb-girdle muscular dystrophy 2B and Miyoshi myopathy
in a Chinese pedigree. Zhonghua Yi Xue Yi Chuan Xue Za Zhi.
21:128–131. 2004.(In Chinese). PubMed/NCBI
|
|
10
|
Bejaoui K, Hirabayashi K, Hentati F,
Haines JL, Ben Hamida C, Belal S, Miller RG, McKenna-Yasek D,
Weissenbach J and Rowland LP: Linkage of Miyoshi myopathy (distal
autosomal recessive muscular dystrophy) locus to chromosome
2p12-14. Neurology. 45:768–772. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bashlr R, Strachan T, Keers S, Stephenson
A, Mahjneh I, Marconi G, Nashef L and Bushby KM: A gene for
autosomal recessive limb-girdle muscular dystrophy maps to
chromosome 2p. Hum Mol Genet. 3:455–457. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Argov Z, Sadeh M, Mazor K, Soffer D,
Kahana E, Eisenberg I, Mitrani-Rosenbaum S, Richard I, Beckmann J,
Keers S, et al: Muscular dystrophy due to dysferlin deficiency in
Libyan Jews Clinical and genetic features. Brain. 123:1229–1237.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Barohn RJ, Amato AA and Griggs RC:
Overview of distal myopathies: From the clinical to the molecular.
Neuromuscul Disord. 8:309–316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Park HJ, Hong JM, Suh GI, Shin HY, Kim SM,
Sunwoo IN, Suh BC and Choi YC: Heterogeneous characteristics of
Korean patients with dysferlinopathy. J Korean Med Sci. 27:423–429.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miyoshi K, Kawai H, Iwasa M, Kusaka K and
Nishino H: Autosomal recessive distal muscular dystrophy as a new
type of progressive muscular dystrophy. Seventeen cases in eight
families including an autopsied case. Brain. 109:31–54. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang M, Zuo YW, Lu Y, Xu M, Liu Z and Jia
JP: Clinical and pathological features of Miyoshi myopathy with
dysferlin protein deficient. Lin Chuang Shen Jing Bing Xue Za Zhi.
22:16–18. 2009.(In Chinese).
|
|
17
|
Zhang LR, Hu J, Zhao Z, Li N, Shen HR and
Bing Q: An analysis of clinical features and pathology in 40
patients with dysferlinopathy. Zhonghua Shen Jing Ge Za Zhi.
46:438–442. 2013.(In Chinese).
|
|
18
|
Ueyama H, Kumamato T, Horinouchi H,
Fujimoto S, Aono H and Tsuda T: Clinical heterogeneity in
dysferlinopathy. Intern Med. 41:532–536. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barohn RJ, Miller RG and Griggs RC:
Autosomal recessive distal dystrophy. Neurology. 41:1365–1370.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aoki M, Liu J, Richard I, Bashir R,
Britton S, Keers SM, Oeltjen J, Brown HE, Marchand S, Bourg N, et
al: Genomic organization of the dysferlin gene and novel mutations
in Miyoshi myopathy. Neurology. 57:271–278. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Galassi G, Rowland LP, Hays AP, Dimauro S
and Hopkins LC: High serum levels of creatine kinase: Asymptomatic
prelude to distal myopathy. Muscle Nerve. 10:346–350. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Linssen WH, Notermans NC, Van der Graaf Y,
Wokke JH, Van Doorn PA, Höweler CJ, Busch HF, De Jager AE and De
Visser M: Miyoshi-type distal muscular dystrophy. Clinical spectrum
in 24 Dutch patients. Brain. 120:1989–1996. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gayathri N, Alefia R, Nalini A, Yasha TC,
Anita M, Santosh V and Shankar SK: Dysferlinopathy: Spectrum of
pathological changes in skeletal muscle tissue. Indian J Pathol
Microbiol. 54:350–354. 2011. View Article : Google Scholar : PubMed/NCBI
|