Introduction
The human sodium taurocholate cotransporting
polypeptide (NTCP) was cloned, localized and functionally
characterized in the year 1994 (1).
NTCP is encoded by the solute carrier family 10 member 1 (SLC10A1)
gene and serves a key role in the enterohepatic circulation of bile
salts, acting as the primary transporter of conjugated bile salts
from the plasma into hepatocytes (2,3).
Although the function of NTCP has been extensively studied and a
number of SLC10A1 genetic variations have been identified in humans
(4–6), the understanding of NTCP deficiencies
remains limited. To date, only one patient with NTCP deficiency has
been diagnosed (7); this patient was
a pediatric patient with the homozygous and pathogenic p.R252H
mutation in the SLC10A1 gene, clinically characterized by marked
conjugated hypercholanemia and otherwise unremarkable
manifestations. The diagnosis of this child patient with NTCP
deficiency clearly established a primary role for NTCP in hepatic
bile acid clearance and advances our understanding of normal
physiology and the disease (8).
However, to the best of our knowledge, no adult patient with NTCP
deficiency has been diagnosed yet so far worldwide.
In the present study, the clinical and molecular
findings of a second child with NTCP deficiency are described, and
an adult with the same genotype and similar, but mild, biochemical
alterations are diagnosed as the first adult case of NTCP
deficiency..
Materials and methods
Subjects and ethical approval
A six-month-old male infant, suspected to have NTCP
deficiency, and his parents participated in the present study in
the First Affiliated Hospital of Jinan University (Guangzhou,
China). Clinical data were collected from the child and analyzed.
Blood samples from 75 healthy volunteers (with a total of 150
SLC10A1 alleles) were collected for allele frequency analysis of
the SLC10A1 variation identified in the patient.
The current study was approved by the Committee for
Medical Ethics (The First Affiliated Hospital, Jinan University,
Guangzhou, China). Written informed consent was obtained from the
parents of the child and from each healthy volunteer.
SLC10A1 gene analysis
To identify mutations in the SLC10A1 gene of the
patient, each of the 5 exons and their flanking sequences,
including the 5′- and 3′-untranslated regions and 309 base pairs
upstream of the transcriptional start site, were amplified by
polymerase chain reaction (PCR) using the primers and polymerases
listed in Table I. All PCR
amplification was conducted using a Mastercycler nexus PCR
instrument (Eppendorf Instrumente GmbH, Hamburg, Germany). SLC25A13
exons 1, 2 and 5 were amplified using a PCR kit (Takara
Biotechnology Co., Ltd., Dalian, China) in a 50-µl mixture that
consisted of 0.25 µl Taq (5 U/µl), 5 µl 10X Buffer (Mg2+
Plus), 1 µl genomic DNA, 4 µl dNTP Mixture (2.5 mM) and 37.75 µl
PCR-grade water, besides the relevant primer pair (1 µl of each
primer in 20 µM). For the LA-PCR amplification of exons 3 and 4,
the components of the reaction mixture was the same as above but
the primer pair and a LA-Taq (Takara Biotechnology Co., Ltd.)
instead of the polymerase Taq. The products were purified using a
gel extraction kit (Omega Bio-Tek, Inc., Norcross, GA, USA), and
analyzed using the Sanger sequencing on a 96-capillary ABI 3730xl
DNA Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Foster City, CA, USA) with a BigDye Terminator v 3.1 Cycle
Sequencing Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA),
according to the manufacturer's instructions.
 | Table I.Polymerase chain reaction primers and
conditions for solute carrier family 10 member 1 gene
sequencing. |
Table I.
Polymerase chain reaction primers and
conditions for solute carrier family 10 member 1 gene
sequencing.
| Exon | NTCP primer
sequence | AT (°C) | Polymerase | Product (bp) |
|---|
| 1 | Forward
5′-GAAACTAAGGAATCAAGAGCGGAGC-3′ | 56 | Taq | 1,248 |
|
| Reverse
5′-CAGGAATTTGAGGTGCTCATTTGG-3′ |
|
|
|
| 2 | Forward
5′-CTTACTACCTTGTGCGACTTTGAG-3′ | 56 | Taq | 987 |
|
| Reverse
5′-GGAATTGGATCTTGTTTCTCTCG-3′ |
|
|
|
| 3–4 | Forward
5′-GTACAAAATGTGGTAGCCTATGGAG-3′ | 56 | LA-Taq | 3,682 |
|
| Reverse
5′-GTTCTCTGGTCTGTCTTGAGGTTC-3′ |
|
|
|
| 5 | Forward
5′-CGAAGTTAGAAGTGAAGTGATGATGAAG-3′ | 58 | Taq | 1,432 |
|
| Reverse
5′-CTGTGTTTCTCGTTTTGGTGTTGG-3′ |
|
|
|
PCR-restriction fragment length
polymorphism (PCR-RFLP) and variation screening
A novel PCR-RFLP procedure was developed for the
present study in order to confirm the genotypes of the patients
family and to screen for variation between the 75 healthy
volunteers. The nucleotide sequences of the forward and reverse
primers used in PCR were 5′-CCAGTTCCCTCTGAGTGTATGTG-3′ and
5′-GCAGGCTCAGGTCTAATATTGG-3′, respectively (Invitrogen; Thermo
Fisher Scientific, Inc.). The HphI restriction enzyme was
used (Thermo Fisher Scientific, Inc.). The PCR temperature profile
was 94°C for 5 min, followed by 35 cycles of 94°C for 30 sec, 60°C
for 40 sec, 72°C for 30 sec, then 72°C for 10 min. To calculate the
variation frequency, the number of alleles bearing the variation in
control subjects was divided by 150 then multiplied by 100.
Alignment of homologous peptides
The peptide identification and amino acid sequences
were collected from the orthologue list of the human SLC10A1 gene
in the Ensembl Genome Browser (www.ensembl.org). The amino acid sequences of 40
homologous peptides in a diversity of species were aligned using
BLAST/BLAT Ensembl software (http://asia.ensembl.org/Multi/Tools/Blast?db=core).
Species included primates (human NTCP-1 and apical sodium-dependent
bile acid transporter, gorilla, orangutan, olive baboon, gibbon,
chimpanzee, macaque, marmoset, vervet African green money and
bushbaby), rodents (opossum, rat, mouse and squirrel),
laurasiatheria (ferret, armadillo, megabat and microbat), placental
mammals (elephant, panda, horse, cow, sheep, alpaca, sloth, tree
shrew, pig, dog, cat, shrew and dolphin), sauropsida (duck,
chicken, flycatcher, softshell turtle and turkey), fish (cave fish
and zebrafish) and invertebrate marine chordate (Ciona
intestinalis).
In silico prediction of
pathogenicity
PolyPhen-2, MutationTaster and SIFT were used to
predict the pathogenicity of the SLC10A1 variation identified in
the patient. PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) analysis
identifies variation as ‘probably damaging’ if the probability is
>0.85, and identifies the variation as ‘possibly damaging’ if
the probability is >0.15 (9).
MutationTaster (http://mutationtaster.org/MutationTaster/index.html)
prediction identifies probabilities ~1 as a ‘high security’ of the
prediction (10). SIFT (http://sift.jcvi.org/www/SIFT_chr_coords_submit.html)
identifies the variation as being ‘deleterious’ if the SIFT score
is <0.05 (11).
Whole genome sequencing
To eliminate the possibility of other genetic causes
resulting in hypercholanemia in the patient, whole genome
sequencing was performed using next generation sequencing (NGS).
Genomic DNA from the patient was extracted from 2 ml peripheral
blood. For quality-control, DNA concentrations were tested with a
Nanodrop Spectrophotometer (Thermo Fisher Scientific, Inc.).
Following quality-control measurements, the DNA was randomly
fragmented into sections of 200–300 base pairs by using a
ultrasonicator (Covaris, Inc., Woburn, MA, USA), then fragments of
the desired length were purified and separated using gel
electrophoresis in a 2% agarose gel, purified with a QIAquick PCR
Purification Kit (QIAGEN, Suzhou, Jiangsu, China), added into End
Repair Mix (New England Biolabs, Inc., Ipswich, MA, USA), and
incubated at 20°C for 30 min. The end-repaired DNA was purified
with a QIAquick PCR Purification Kit (QIAGEN), then added to an
A-Tailing Mix (New England Biolabs, Inc.), and incubated at 37°C
for 30 min. Then, a ligation reaction was conducted by incubating
the purified Adenylate 3′ Ends DNA, Adapter and Ligation Mix (New
England Biolabs, Inc.) at 20°C for 15 min. Adapter-ligated DNA was
selected by running a 2% agarose gel and purified with a QIAquick
Gel Extraction kit (QIAGEN). Following that, several rounds of PCR
amplification were performed using a PCR Primer Cocktail and PCR
Master Mix (New England Biolabs, Inc.) to enrich the
Adapter-ligated DNA fragments. Then the PCR products were selected
by running another 2% agarose gel and purified with a QIAquick Gel
Extraction kit (QIAGEN). Adapter ligation and DNA cluster
preparation were performed and the DNA fragments were analyzed
using the Illumina Hiseq 2000 Sequencing System (Illumina, Inc.,
San Diego, CA, USA). A total of 95.92 G clean databases were
generated for the DNA library that was constructed for the DNA
sample. To ensure high quality analysis, the adapter pollutions and
the reads which contained ≥50% low quality bases (quality value,
≤5) were removed, ensuring that the Q20 base rate of each lane was
>93.77%. The sequencing reads were aligned onto the reference
genome sequence using the Short Oligonucleotide Analysis Package
aligner/soap2 (http://soap.genomics.org.cn/soapaligner.html; BGI,
Shenzhen, China) and single nucleotide polymorphisms (SNPs), InDels
and structure variations (SVs) of the sequenced genome were
analyzed, as described below.
To annotate and prioritize variation within the
gene, common intronic variants were removed following comparison
with those in the Single Nucleotide Polymorphism Database and the
1000 Genomes Project (National Center for Biotechnology
Information, Bethesda, MD, USA). Synonymous and non-synonymous
mutations in exons that were not deleterious following in
silico prediction using PolyPhen-2, SIFT or MutationTaster,
were also deleted. The variants that were causing abnormal splicing
or amino acid changes (including stop-loss and stop-gain variants),
in particular those with minor allele frequencies (MAFs) <1%,
were recorded as possible candidates of the causative mutations. In
view of the intractable and marked hypercholanemia observed in the
patient, a panel of genes that may uniquely affect bile acid
homeostasis were assembled for candidate mutation analysis using
PubMed, The Online Mendelian Inheritance in Man record and
primarily by reference to previous publications (12–16).
In summary, variation pathogenicity was analyzed
using in silico methods of prediction using a number online
tools. Next, the phenotypic features, including the genetic pattern
of the genes involved in harboring pathogenic variations, were
compared against the clinical and laboratory findings in the
patient. Variations that were identified as causing cholestasis or
affecting bile acid homeostasis were considered as being involved
in affecting the phenotype of the patient.
Results
Case report
A 6-month-old male infant was admitted to the First
Affiliated Hospital of Jinan University as a result of elevated
serum total bile acid (TBA) discovered 4 months previously. At the
age of 2 months, the patient underwent a liver function test due to
mild jaundice, and it was observed that the levels of TBA and
direct bilirubin were elevated. No steatorrhea or acholic stool was
observed. After 1 month, the direct bilirubin level recovered
gradually; however, the TBA remained elevated for 4 months. The
infant was delivered by cesarean section at the gestation age of 38
weeks and 2 days (weight, 3.6 kg; body length, 50 cm) due to
entanglement of the umbilical cord. The infant's parents were
clinically and biochemically healthy with no family history of
genetic disease.
The patient underwent a physical examination at 6
months (body weight, 8.5 kg; height, 71 cm; head circumference,
44.5 cm) and no dysmorphic appearance or jaundice was observed in
the skin and sclera. No stridor, crackles or crepitus was heard
with auscultation of the two lungs. The heart sound was normal
without any murmurs. The liver and spleen were non-palpable. When
picking the infant up, the tone of the body and limbs appeared
normal. On biochemical analysis, the levels of TBA reached 492.8
µmol/l (0–10 µmol/l), whereas other indices were normal (Table II). Since citrin deficiency is a
common etiology of infant cholestasis in Chinese, genetic analysis
of the causative gene, SLC25A13, was performed; however, no
mutations were observed, thus eliminating the possibility of citrin
deficiency.
| Table II.Biochemical indices over time in a
patient with marked hypercholanemia. |
Table II.
Biochemical indices over time in a
patient with marked hypercholanemia.
|
|
Months |
|---|
|
|
|
|---|
| Indices (reference
range) | 2 | 3 | 4 | 5 | 6a | 8.5 | 13b | 19 | 24.5 | 30.5 |
|---|
| ALT (5–40 U/l) | 33 | 30 | 37 | 31 | 32 | 34 | 27 | 24 | 16 | 17 |
| AST (5–40 U/l) | 49 | 32 | 37 | 38 | 37 | 36 | 41 | 37 | 31 | 36 |
| GGT (8–50 U/l) | 220 | 68 | 28 | 11 | 11 | 8 | 9 | 9 | 9 | 9 |
| ALP (20–500
U/l) | 383 | 345 | 317 | 273 | 236 | 332 | 270 | 248 | 254 | 247 |
| Tbil (2–19
µmol/l) | 133.5 | 30.5 | 10.9 | 7.2 | 9.6 | 9.4 | 8.4 | 7.3 | 12.0 | 9.3 |
| Dbil (0–6
µmol/l) | 98.4 | 23.0 | 7.6 | 4.5 | 6.0 | 4.8 | 4.7 | 4.2 | 5.8 | 4.9 |
| Ibil (2.56–20.9
µmol/l) | 35.1 | 7.5 | 3.3 | 2.7 | 3.5 | 4.6 | 3.7 | 3.1 | 6.2 | 4.4 |
| TBA (0–10
µmol/l) | 221.9 | 151.4 | 431.4 | 251.5 | 492.8 | 567.8 | 653.1 | 494.5 | 737.8 | 405.8 |
During the subsequent 2 years, itchy maculopapular
rashes occurred occasionally in the trunk and extremities, and
these were attributed to mite dermatitis. This conclusion was
reached as serum IgE reached 1,460 IU/ml (0–60), the serum antigens
of Dermatophagoides pteronyssinus and Dermatophagoides
farinae were strongly positive, and anti-mite measures were
able to effectively relieve the severity and frequency of the rash.
His TBA reached a surprisingly high level of 737.8 µmol/l at the
age of 24.5 months (Table II).
Therefore, it was concluded that the pruritus was allergic, not
cholestatic, in the patient. Although a low serum level of total
25-OH vitamin D [21.4 ng/ml (30–100)] at the age of 2 years and 7
months indicated vitamin D insufficiency, no obvious clinical
presentations of fat soluble vitamin malabsorption were observed.
The patient's anthropometric indices and social performance
developed well without any symptoms or signs of hypotonia, delayed
motor milestones and growth retardation on clinical follow-up.
However, the hypercholanemia was marked and intractable, regardless
of the introduction of cholestyramine at the age of 13 months.
Therefore, at 30.5 months, SLC10A1 analysis was performed to
evaluate the possibility of NTCP deficiency.
SLC10A1 genotypes and variation
frequency
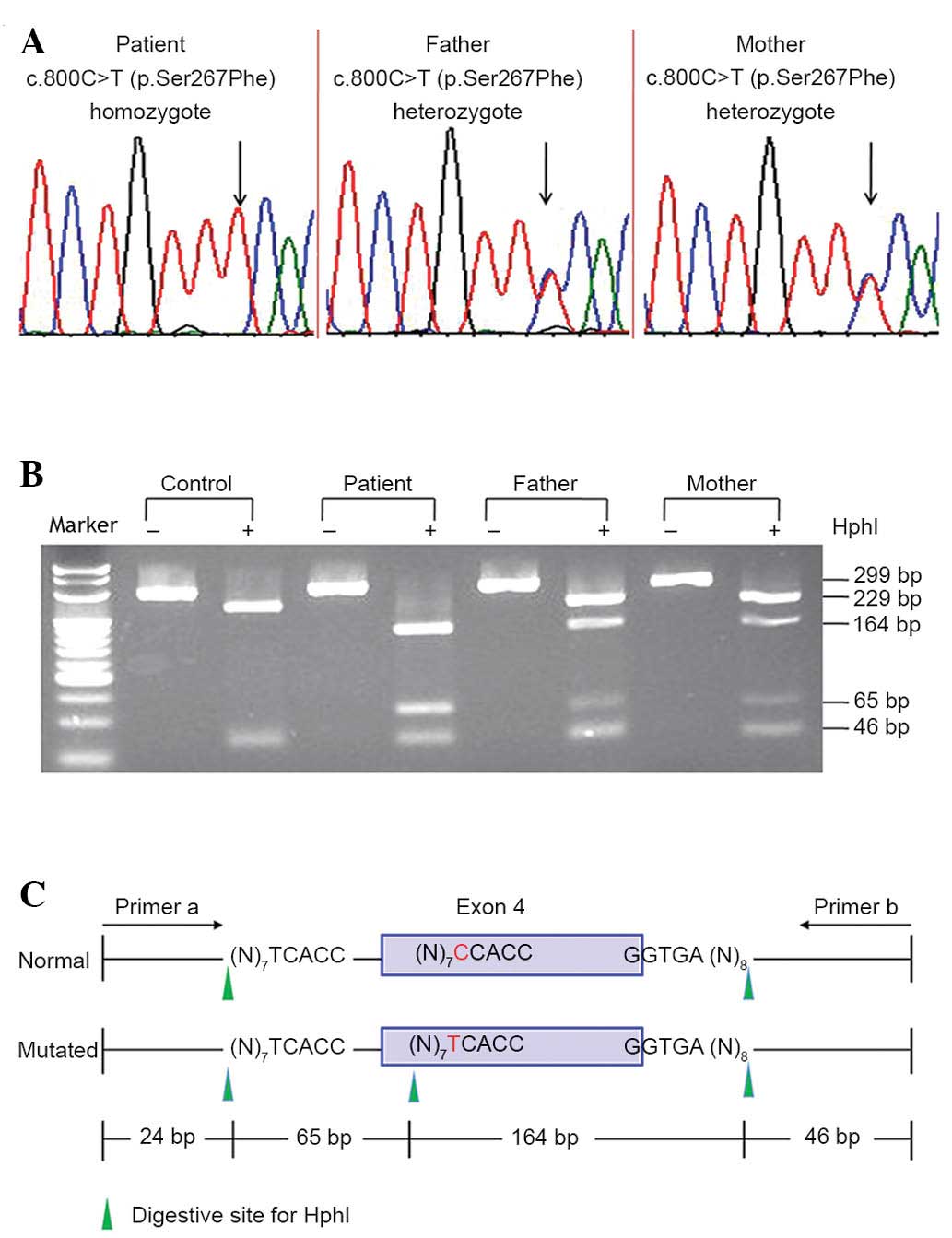
Sanger sequencing of the SLC10A1 gene demonstrated
that the patient was a homozygote of the variation c.800C>T
(p.Ser267Phe), and that both parents were carriers of the
variation. A novel RFLP procedure was developed that confirmed the
SLC10A1 genotype of the patient and his parents (Fig. 1). Screening for the p.Ser267Phe
variation in the 75 control subjects was performed using the RFLP
procedure, revealing 5 heterozygous carriers and 1 homozygote, who
was a 30-year-old female without any positive symptoms or signs of
NTCP but with a slightly elevated level of TBA [19.3 µmol/l (0–10
µmol/l)]. A 4.7% (7/150) allele frequency within the 75 volunteers
indicated that the p.Ser267Phe variation is an SLC10A1 SNP.
Bioinformatics
As presented in Fig.
2, the amino acid residue p.267Ser in human NTCP is relatively
conserved among aligned homologous peptides from a number of
species. SIFT analysis indicated that the c.800C>T (p.Ser267Phe)
variation was predicted to be ‘deleterious’ (SIFT score, 0.01).
Using PolyPhen-2 software, a probability score of 0.959 was
calculated, indicating that the variation is ‘probably damaging’.
Meanwhile, MutationTaster evaluation concluded that the variation
is ‘disease-causing’ (probability value, 0.911).
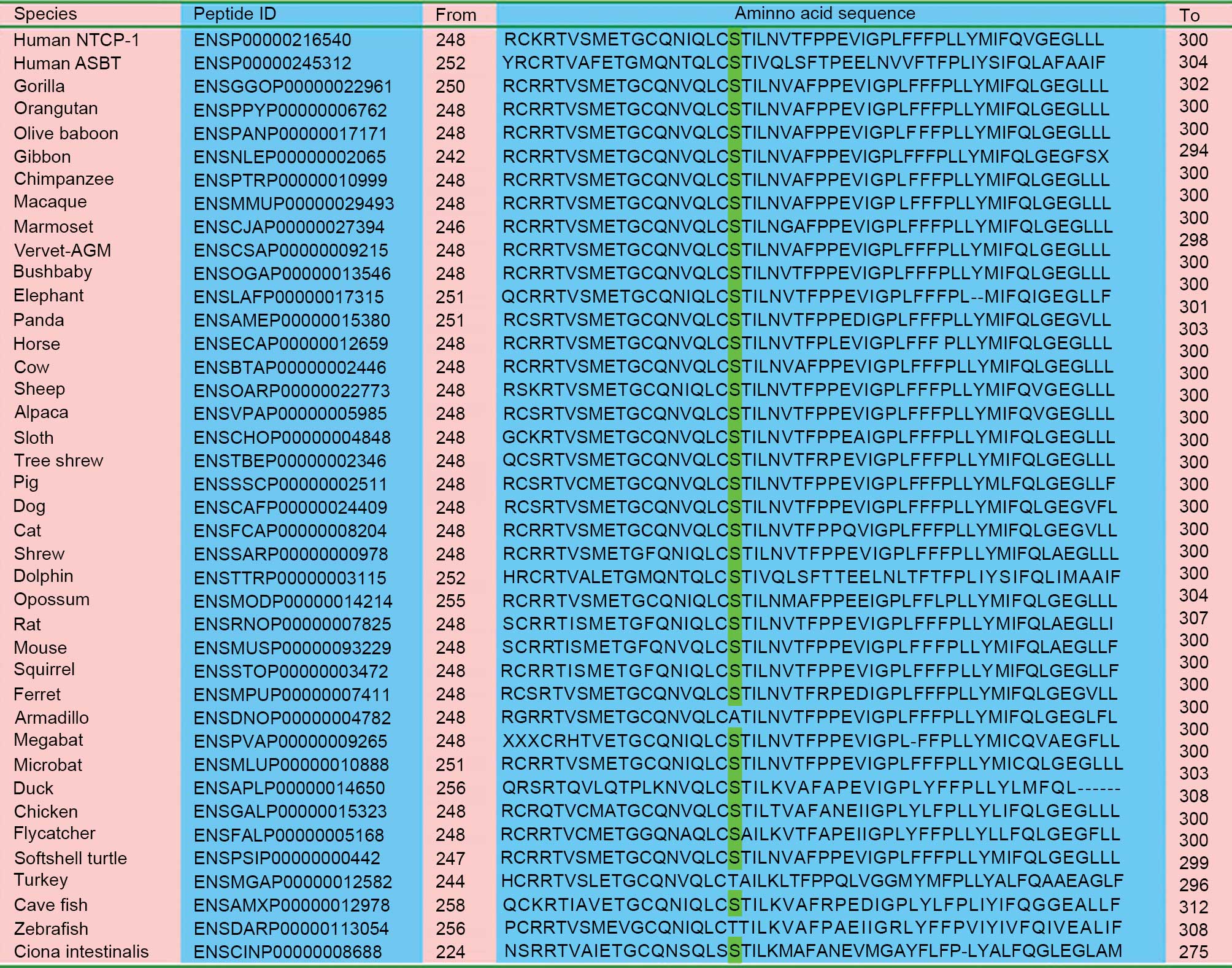 | Figure 2.Alignment of homologous peptides in a
diversity of species. With the exception of armadillo, turkey and
zebrafish, all the remaining 37 species, including primates,
rodents, laurasiatheria, placental mammals, sauropsida, fish and
marine chordate, have the same serine residue as highlighted in
green This indicates that the p.Ser267Phe variation affected a
number of conserved amino acid residues among species. NTCP, sodium
taurocholate cotransporting polypeptide; ASBT, apical
sodium-dependent bile acid transporter. |
NGS results
NGS analysis revealed 85 splicing, 80
stop-gain/stop-loss and 10,490 exonic nonsynonymous SNPs, and 586
exonic indels and 76 exonic SVs were detected. Among the NGS
analysis results, 239 exonic nonsynonymous SNPs and 4 stop-gains
with MAF <1% were detected that were ‘benign’ or ‘tolerated’ on
functional prediction, and not associated with the clinical and
biochemical features of the patient.
The bioinformatic tools PolyPhen-2, MutationTaster
and SIFT, did not detect any ‘deleterious’ or ‘damaging’ variations
among the NGS data of the 93 genes which were associated with
inherited infantile cholestatic disorders (16). When focusing on the 51 genes that
affect bile acid homeostasis uniquely, only 12 exonic nonsynonymous
SNPs were detected (Table III).
Aside from the p.S267F variation (rs2296651), none of the
variations were predicted, clinically or bioinformatically, to be
the causative variation of hypercholanemia in the patient.
Consequently, the biallelic dysfunctional p.S267F variation in the
SLC10A1 gene was concluded to be the unique determinant causing
NTCP deficiency in the patient.
| Table III.Nonsynonymous exonic SNPs detected in
genes that may affect bile acid homeostasis. |
Table III.
Nonsynonymous exonic SNPs detected in
genes that may affect bile acid homeostasis.
| No. | Gene | OMIM ID | Location | dbSNP detected | Amino acid
changes | Status |
|---|
| 01 | ATP8B1 | 602397 | 18q21.31 |
|
|
|
| 02 | ABCB11 | 603201 | 2q31.1 |
rs2287622 |
p.V444A | Heterozygous |
| 03 | TJP2 | 607709 | 9q21.11 |
|
|
|
| 04 | VPS33B | 608552 | 15q26.1 |
rs11073964 |
p.G487S | Homozygous |
| 05 | VIPAS39 | 613401 | 14q24.3 |
|
|
|
| 06 | CCBE1 | 235510 | 18q21.32 |
|
|
|
| 07 | AMACR | 604489 | 5p13.2 |
rs3195676 |
p.V9M | Heterozygous |
| 08 | CYP7A1 | 118455 | 8q12.1 |
|
|
|
| 09 | AKR1D1 | 604741 | 7q33 |
|
|
|
| 10 | HSD3B7 | 607764 | 16p11.2 |
|
|
|
| 11 | CYP7B1 | 603711 | 8q12.3 |
|
|
|
| 12 | CYP27A1 | 606530 | 2q35 |
|
|
|
| 13 | SLC27A5 | 603314 | 19q13.43 |
|
|
|
| 14 | BAAT | 602938 | 9q31.1 |
|
|
|
| 15 | HSD17B4 | 601860 | 5q23.1 |
rs11205 |
p.I541V | Homozygous |
| 16 | SLC25A13 | 603859 | 7q21.3 |
|
|
|
| 17 | SERPINA1 | 107400 | 14q32.13 |
|
|
|
| 18 | BCS1L | 603647 | 2q35 |
|
|
|
| 19 | ABCB4 | 171060 | 7q21.12 |
|
|
|
| 20 | CLDN1 | 603718 | 3q28 |
|
|
|
| 21 | JAG1 | 601920 | 20p12.2 |
|
|
|
| 22 | NOTCH2 | 600275 | 1p12-p11 |
|
|
|
| 23 | CFTR | 602421 | 7q31.2 |
|
|
|
| 24 | Cirhin | 604901 | 16q22.1 |
|
|
|
| 25 | SLC10A2 (ABST) | 613291 | 13q33.1 |
rs188096 |
p.S171A | Heterozygous |
| 26 | EPHX1 | 132810 | 1q42.12 |
|
|
|
| 27 | NTCP (SLC10A1) | 182396 | 14q24.2 |
rs2296651 |
p.S267F | Homozygous |
| 28 | ABCC2 (MRP2) | 237500 | 10q24.2 |
rs17222589 |
|
|
| 29 | SLCO1B3 | 605495 | 12p12.2 |
|
|
|
| 30 | SLCO1B1 | 604843 | 12p12.2-p12.1 |
|
|
|
| 21 | MRP6 (ABCC6) | 603234 | 16p13.11 |
|
|
|
| 32 | ABCG5 | 605459 | 2p21 |
rs6756629 |
p.R50C | Heterozygous |
| 33 | ABCG8 | 605460 | 2p21 |
rs6544718 |
p.V632A | Homozygous |
| 34 | SLCO2B1 | 604988 | 11q13.4 |
rs2306168 |
p.S342F | Heterozygous |
| 35 | SLCO1A2 | 602883 | 12p12.1 |
|
|
|
| 36 | SLCO4A1 | 612436 | 20q13.33 |
|
|
|
| 37 | SLC51A (OSTα) | 612084 | 3q29 |
|
|
|
| 38 | SLC51B (OSTβ) | 612085 | 15q22 |
|
|
|
| 39 | TMEM30A | 611028 | 6q14.1 |
|
|
|
| 40 | TMEM30B | 611029 | 14q23.1 |
|
|
|
| 41 | TMEM30C | 611030 | 3q12.1 |
|
|
|
| 42 | NR1H4 | 603826 | 12q23.1 |
|
|
|
| 43 | CYP8B1 | 602172 | 3p22.1 |
rs9865715 |
p.S88P | Homozygous |
| 44 | SLC4A2 | 109280 | 7q36.1 |
|
|
|
| 45 | SLC10A7 | 611459 | 4q31.2 |
|
|
|
| 46 | SLC22A1
(OCT-1) | 602607 | 6q25.3 |
rs628031 |
p.M408V | Homozygous |
| 47 | SLC22A7
(OAT-2) | 604995 | 6p21.1 |
|
|
|
| 48 | MDR1 (ABCB1) | 171050 | 7q21.12 |
|
|
|
| 49 | MRP3 (ABCC3) | 604323 | 17q21.33 |
|
|
|
| 50 | MRP4 (ABCC4) | 605250 | 13q32.1 |
|
|
|
| 51 | SLC47A1
(MATE-1) | 609832 | 17p11.2 |
|
|
|
Discussion
Vaz et al (7)
first reported a patient with NTCP deficiency, who presented with
extremely elevated TBA levels (≤1,500 µM, reference range, <16.3
µM) but otherwise a relatively mild clinical phenotype
characterized by mild hypotonia, growth retardation and delayed
motor milestones. The patient in the present case report presented
with intractable and marked hypercholanemia with normal
anthropometric development and social performance. The unique
disturbance in bile salt homeostasis suggested that NTCP was the
most likely affected molecule in the patient. A homologous
p.Ser267Phe variation in the SLC10A1 gene was detected using Sanger
sequencing, and orthologue alignment and pathogenicity prediction
supported the disease-causing nature of this variation. In
addition, the p.Ser267Phe variant was reported to exhibit a near
complete loss of function of bile acid uptake, causing NTCP to lose
its taurocholate transporting activity (5,17).
The experimental findings in the present study were
similar to the functional analysis of the causative mutation
p.Arg252His in the first patient that was reported to have NTCP
deficiency, who was a homozygote of the causative mutation
p.Arg252His (7). The p.Ser267Phe and
p.Arg252His variations demonstrated similarly strong experimental
and bioinformatic pathogenicity (5,7,17); using various in silico tools,
the p.Arg252His mutation displayed a disease-causing nature with a
score of 1.000 using PolyPhen-2, 0.9994 using MutationTaster and 0
using SIFT analysis. These results are similar to the functional
predictions of the p.Ser267Phe SNP in the current study. Together,
this evidences supports the diagnosis of NTCP deficiency in the
patient in the present study.
As the genetic determinants of hypercholanemia are
complex (16), whole genome
sequencing was performed in the present study in order to evaluate
the possible involvement of other genes, the collective testing of
which was prohibitively time-consuming and cost-expensive. The
sequencing revealed that no other genetic mutations were causing
perturbations of bile acid metabolism other than the SLC10A1 gene,
and this further supports the diagnosis of NTCP deficiency in the
patient. To the best of our knowledge, this is the second report of
a patient with NTCP deficiency in the world.
The allele frequency of the p.Ser267Phe variation
was calculated to be 4.7% in control subjects. This is consistent
with previous studies which demonstrated that this polymorphism is
common in East Asian countries including China and Vietnam, but not
in European American, African American or Hispanic countries
(4–6,18). NTCP
is the functional receptor for human hepatitis B and D viruses
(19). The p.Ser267Phe variation
experimentally abrogates the ability of NTCP to support HBV
infections in cell culture (17),
and is associated with resistance to chronic hepatitis B in humans
(18). This supports the concept
that p.Ser267Phe SNP severely impairs the function of NTCP, and
suggests that its high allele frequency may be a result of positive
selection in Eastern Asia, where hepatitis B is more prevalent than
in European American, African American or Hispanic countries
(18).
Besides the NTCP-deficient biallelic p.Ser267Phe in
the child patient in the present study, an adult homologous for the
same dysfunctional SLC10A1 SNP was identified in the present study,
who presented with slight hypercholanemia without other clinical or
laboratory anomalies. The mechanism underlying the more prominent
hypercholanemia in the infant, in comparison with the adult with
the same SLC10A1 genotype, remains unknown. It is likely that the
adult homozygote of the p.Ser267Phe SNP had, or lacked, a number of
other genomic SNPs that were analyzed in the child (Table III). Another possible explanation
is that the impaired NTCP function in the adult may have been
compensated for by other transporters with the ability to uptake
bile acids from the plasma, such as Organic Anion Transporting
Polypeptide (OATP) 1B1 and OATP1B3 in the basolateral membrane of
hepatocytes (20), and the type II
form of microsomal epoxide hydrolase (21).
In addition to the prominent hypercholanemia, the
elevated serum levels of ALT, AST, TBil and DBil in the child
(Table II) suggested the presence
of transient cholestasis in early infancy. The increased levels of
GGT within the first 3 months after birth may have been partially
attributed to cholestasis, although GGT activity is typically
higher in infants than in adults (22). As the child was delivered by cesarean
section due to entanglement of the umbilical cord, hypoxic ischemia
of the liver may have been a likely cause for the transient
elevation of the cholestatic indices. Another possibility is that
the transient cholestatic liver disease experienced in early
infancy may be a phenotypic feature of NTCP deficiency in the
child. However, further research is required to determine whether
liver disease is common in pediatric patients with NTCP
deficiency.
In summary, the current study presents a pediatric
patient who demonstrated prominent hypercholanemia with otherwise
unremarkable presentations. SLC10A1 gene analysis revealed the
presence of a p.Ser267Phe homozygote that is bioinformatically
analyzed as a dysfunctional SNP that prevents the functioning of
NTCP. Combined with the results from an adult with mild
hypercholanemia, the diagnosis of the child with NTCP deficiency
suggests, for the first time, that there is an association between
the dysfunctional SNP and hypercholanemia. To the best of our
knowledge, this is the second clinical description on NTCP
deficiency worldwide.
Acknowledgements
The authors thank Dr Qun-Zhou Zhang for the revision
of the manuscript. The present study was supported by the National
Natural Science Foundation of China (grant nos. 81270957 and
81570793) and the Cultivation Foundation for Scientific Research
(grant no. 2014208) approved by the First Affiliated Hospital,
Jinan University.
Glossary
Abbreviations
Abbreviations:
|
NTCP
|
sodium taurocholate cotransporting
polypeptide
|
|
RFLP
|
restriction fragment length
polymorphism
|
|
SNP
|
single nucleotide polymorphism
|
|
NGS
|
next generation sequencing
|
|
MAF
|
minor allele frequency
|
|
SVs
|
structural variations
|
|
ALT
|
alanine transaminase
|
|
AST
|
aspartate transaminase
|
|
GGT
|
gamma-glutamyl transpeptidase
|
|
ALP
|
alkaline phosphatase
|
|
TBA
|
total bile acids
|
References
|
1
|
Hagenbuch B and Meier PJ: Molecular
cloning, chromosomal localization, and functional characterization
of a human liver Na+/bile acid cotransporter. J Clin
Invest. 93:1326–1331. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hagenbuch B and Dawson P: The sodium bile
salt cotransport family SLC10. Pflugers Arch. 447:566–570. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Anwer MS and Stieger B: Sodium-dependent
bile salt transporters of the SLC10A transporter family: More than
solute transporters. Pflugers Arch. 46:77–89. 2014. View Article : Google Scholar
|
|
4
|
Saito S, Iida A, Sekine A, Ogawa C,
Kawauchi S, Higuchi S and Nakamura Y: Catalog of 238 variations
among six human genes encoding solute carriers (hSLCs) in the
Japanese population. J Hum Genet. 47:576–584. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ho RH, Leake BF, Roberts RL, Lee W and Kim
BB: Ethnicity-dependent polymorphism in Na+-taurocholate
cotransporting polypeptide (SLC10A1) reveals a domain critical for
bile acid substrate recognition. J Biol Chem. 279:7213–7222. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pan W, Song IS, Shin HJ, Kim MH, Choi YL,
Lim SJ, Kim WY, Lee SS and Shin JG: Genetic polymorphisms in
Na+-taurocholate co-transporting polypeptide (NTCP) and
ileal apical sodium-dependent bile acid transporter (ASBT) and
ethnic comparisons of functional variants of NTCP among Asian
populations. Xenobiotica. 41:501–510. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vaz FM, Paulusma CC, Huidekoper H, de Ru
M, Lim C, Koster J, Ho-Mok K, Bootsma AH, Groen AK, Schaap FG, et
al: Sodium taurocholate cotransporting polypeptide (SLC10A1)
deficiency: Conjugated hypercholanemia without a clear clinical
phenotype. Hepatology. 61:260–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karpen SJ and Dawson PA: Not all (bile
acids) who wander are lost: the first report of a patient with an
isolated NTCP defect. Hepatology. 61:24–27. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boyer JL: Bile formation and secretion.
Compr Physiol. 3:1035–1078. 2013.PubMed/NCBI
|
|
13
|
Hirschfield GM: Genetic determinants of
cholestasis. Clin Liver Dis. 17:147–159. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marschall HU and Beuers U: When bile acids
don't get amidated. Gastroenterol. 144:870–873. 2013. View Article : Google Scholar
|
|
15
|
Sambrotta M, Strautnieks S, Papouli E,
Rushton P, Clark BE, Parry DA, Logan CV, Newbury LJ, Kamath BM,
Ling S, et al: Mutations in TJP2 cause progressive cholestatic
liver disease. Nat Genet. 46:326–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Herbst SM, Schirmer S, Posovszky C, Jochum
F, Rödl T, Schroeder JA, Barth TF, Hehr U, Melter M and Vermehren
J: Taking the next step forward - Diagnosing inherited infantile
cholestatic disorders with next generation sequencing. Mol Cell
Probes. 9:291–29. 2015. View Article : Google Scholar
|
|
17
|
Yan H, Peng B, Liu Y, Xu G, He W, Ren B,
Jing Z, Sui J and Li W: Viral entry of hepatitis B and D viruses
and bile salts transportation share common molecular determinants
on sodium taurocholate cotransporting polypeptide. J Virol.
88:3273–3284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng L, Zhao Q, Li Q, Li M, Li C, Xu T,
Jing X, Zhu X, Wang Y, et al: The p. Ser267Phe variant in SLC10A1
is associated with resistance to chronic hepatitis B. Hepatology.
61:1251–1260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z,
Huang Y, Qi Y, Peng B, Wang H, et al: Sodium taurocholate
cotransporting polypeptide is a functional receptor for human
hepatitis B and D virus. eLife. 1:e000492012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hagenbuch B and Meier PJ: The superfamily
of organic anion transporting polypeptides. Biochim Biophys Acta.
1609:1–18. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu QS, Xing W, Qian B, von Dippe P,
Shneider BL, Fox VL and Levy D: Inhibition of human m-epoxide
hydrolase gene expression in a case of hypercholanemia. Biochim
Biophys Acta. 1638:208–216. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cabrera-Abreu JC and Green A:
Gamma-glutamyltransferase: Value of its measurement in paediatrics.
Ann Clin Biochem. 39:22–25. 2002. View Article : Google Scholar : PubMed/NCBI
|