Introduction
Ovarian cancer is the fifth most common cause of
death in women worldwide (1).
Typical treatment for ovarian cancer involves cytoreductive surgery
combined with cisplatin-based chemotherapy (2). Unfortunately, the initial response rate
of this treatment is not sustainable and in 70% of women disease
reoccurs within months as a result of chemoresistance (3,4). It has
been suggested that the high mortality rate in ovarian cancer can
be partly attributed to this high frequency of chemoresistance
(5).
Cisplatin is a platinum compound that was found to
arrest binary fission in Escherichia coli in the 1960s, and
it is now a crucial chemotherapeutic drug in the treatment of
numerous cancers, including ovarian, as a single agent and in
combination with other anticancer drugs (6,7). The
issue of resistance to cisplatin remains a major obstacle in the
successful treatment of ovarian cancer (8). Previous studies have indicated that
inhibition of intrinsic cell death signaling pathways, activation
of cell survival signaling pathways, and dysregulation of
oncogenes, tumor suppressor genes and microRNAs contributes to
cisplatin chemoresistance in ovarian cancer cells (9,10).
However, the underlying mechanisms by which cisplatin
chemoresistance occurs in ovarian cancer cells remain unclear.
Previous studies have identified that abnormal Wnt
signaling serves a role in the development of breast (11), gastric (12), lung (13), prostate (14) and endometrial (15) cancers. In ovarian cancer, persistent
activation of Wnt signaling was observed to increase cell survival,
and several studies have concluded that aberrant regulation of Wnt
signaling induces tumor cell chemoresistance (16–18).
The Wnt signaling signaling pathway serves a
critical role in embryogenesis and oncogenesis (19). Wnt ligands are a family of secreted
proteins comprising of 19 members (10). Wnt ligands bind to a Frizzled (Fzd)
family receptor and low-density lipoprotein receptor-related
protein (LRP)-5/6 to initiate signaling (10). Wnt signaling is divided into
canonical Wnt signaling pathway (Wnt/β-catenin) and non-canonical
Wnt signaling pathways (19).
In the canonical Wnt signaling pathway, illustrated
in Fig. 1A, Wnt ligands engage Fzd
and LRP-5/6, which inhibits glycogen synthase kinase 3β (GSK-3β),
leading to stabilization and increasing expression levels of
β-catenin in the cytoplasm. Stable β-catenin translocates into the
nucleus where it binds to the transcription factor T-cell
factor/lymphoid enhancer-binding factor (TCF/LEF) to control the
transcription of Wnt target genes (20).
 | Figure 1.Illustration of the Wnt signaling
pathways. (A) Wnt/β-catenin signaling pathway. Wnt ligands bind to
Fzd/LRP-5/6, increasing stabilization and accumulation of
β-catenin. β-catenin associates with TCF/LEF to control expression
of target genes. (B) Wnt/Ca2+ signaling pathway. Wnt
activates intracellular Ca2+ release and
Ca2+-dependent protein kinases, such as CaMKII. TAK1 and
NLK interfere with β-catenin/TCF signaling pathway. (C) Wnt/JNK
signaling pathway. Receptor stimulation activates DVL, resulting in
the activation of Rho family GTPases. Rho triggers c-Jun expression
through phosphorylation of JNK. FZD, frizzled; LRP, low-density
lipoprotein receptor-related protein-5/6; DVL, dishevelled; GSK3β,
glycogen synthase kinase 3β; APC, adenomatous polyposis coli; CK1,
casein kinase 1; TCF/LEF, transcription factor/lymphoid
enhancer-binding factor; CaMKII,
Ca2+/calmodulin-dependent protein kinase II; TAK1,
TGF-β-activated kinase 1; NLK, serine/threonine protein kinase NLK;
MEK MAPK/ERK kinase; JNK, c-Jun N-terminal kinase. |
Dysregulation of the Wnt/β-catenin signaling pathway
has been identified in numerous cancers, including ovarian
(21). β-catenin is the primary
component of this signaling pathway, and mutations in the gene
encoding β-catenin (CTNNB1), leading to alteration of the
Wnt/β-catenin signaling pathway, have been found in the
endometrioid subtype of ovarian cancer (22,23).
Furthermore, aberrant expression of β-catenin and Wnt-5A has been
observed in ovarian cancer (22,24). In
addition, aberrant accumulation of β-catenin is associated with
increasing ovarian cancer grade and poor survival (25,26).
However, the use of β-catenin as a prognostic marker for ovarian
cancer is disputed. To the best of our knowledge, the impact of
β-catenin expression levels on chemoresistance has not been
evaluated in ovarian cancer.
The non-canonical Wnt signaling pathways involve
signaling that uses downstream effectors, without mediation by
β-catenin-TCF/LEF. In contrast to canonical Wnt signaling, these
signaling pathways may have transcriptional and non-transcriptional
effects (27). In the non-canonical
Wnt/Ca2+ signaling pathway, Wnt ligands binding to Fzd
receptors initiates the activation of phospholipase C via G
protein-couple receptor signaling, causing an increase in
intracellular Ca2+, resulting in activation of
Ca2+/calmodulin-dependent kinase II (CaMKII) and protein
kinase C. Previous studies have identified that deregulation of the
Wnt/Ca2+ signaling pathway mediates cytoskeleton
rearrangements, cellular proliferation, cellular motility and
epithelial-mesenchymal transition in cancer development and
progression (Fig. 1B) (27).
In the non-canonical Wnt/JNK signaling pathway,
Wnt-Fzd binding activates a number of small
guanosine-5′-triphosphate (GTP) -binding proteins, including c-Jun
N-terminal kinase (JNK), which affect a wide range of cellular
processes, including cytoskeleton rearrangement, cell polarity,
cell migration and gene expression (28). Previous studies have identified that
aberrant Wnt/JNK signaling may initiate and stimulate the
development of malignant phenotypes through effects on cell
proliferation, survival, polarity, invasion and metastasis
(Fig. 1C) (29,30).
Aberrant Wnt/β-catenin, Wnt/JNK and
Wnt/Ca2+ signaling has been associated with increasing
ovarian cancer grade and poor survival (21,27,29).
However, to the best of our knowledge, the relationship between
these signaling pathways and ovarian cancer cisplatin
chemoresistance has not yet been investigated (23,29).
The present study aims to determine the expression
of β-catenin, JNK and CaMKII in a cisplatin-sensitive ovarian
cancer cell line and a cisplatin-resistant variant, and then assess
the correlation between expression of these proteins and cisplatin
chemoresistance.
Materials and methods
Chemicals and reagents
Cisplatin was purchased from Sigma-Aldrich (Merck
Millipore, Darmstadt, Germany). Cell counting kit-8 (CCK-8; C0037)
and 4′,6-diamidino-2-phenylindole (DAPI; C1005) were purchased from
the Beyotime Institute of Biotechnology (Haimen, China) and stored
at −20°C. Antibodies including non-phospho (active) β-catenin
(Ser33/37/Thr41) (D13A1) rabbit mAb (#8814), SAPK/JNK rabbit mAb
(#9252) and CaMKII (pan) (D11A10) Rabbit mAb (#4436) were purchased
from Cell Signaling Technology, Inc. (Beverly, MA, USA), and the
β-actin mouse mAb (JT9001S) was purchased from Anbo Biotechnology
Co., Ltd, (San Francisco, CA, USA). The Wnt signaling inhibitor
(XAV-939) and activator (CHIR-99021) were purchased from the
Selleck Chemicals (Shanghai, China), and completely dissolved in
DMSO at a stock concentration (1 mM). Fluorescein isothiocyanate
(FITC)-Conjugated AffiniPure Goat Anti-Mouse IgG (H+L) (ZF-0312)
and FITC-Conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) (ZF-0315)
were from OriGene Technologies, Inc. (Beijing, China).
Cell culture
The human ovarian cancer adenocarcinoma cell line
SKOV3 and its cisplatin-resistant variant, SKOV3/DDP, were
purchased from The Chinese Academy of Sciences (Shanghai, China).
Cells were cultured in Roswell Park Memorial Institute (RPMI)-1640
media, supplemented with 100 U/ml penicillin/streptomycin and 10%
fetal bovine serum (FBS) at 37°C and 5% CO2. In
addition, the SKOV3/DDP cell media included 2 µM cisplatin to
maintain chemoresistance. All of the described reagents were
purchased from GE Healthcare Life Sciences (HyClone; Logan, UT,
USA).
Cell viability and cytotoxicity
assay
Cell viability was detected using the CCK-8 kit.
Briefly, cells were seeded at 5×103 cells/well into
96-well plates. Following 48 h incubation as same as previously
described conditions, cells were treated with cisplatin,
CHIR-99021, XAV-939, cisplatin + CHIR-99021 or cisplatin + XAV-939
for indicated concentrations. There were six replicate wells for
each concentration, the control group contained untreated cells and
in the blank group (no cells or drugs) only added 10% FBS and
RMPI-1640. Cells were incubated for 24, 48 or 72 h prior to the
addition of 10 µl CCK-8 reagent and 90 µl RMPI-1640 to each well
and further culture for 1 h. The absorbance of each well at 490 nm
(A490) was then analyzed using a microplate reader
(Model 680; Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
effect of the drugs on the cell survival was calculated using the
following formula: Survival rate (%) = [(A490 of treated
group - A490 of blank group) / (A490 of
control group - A490 of blank group)] × 100. The
inhibition rate (%) was calculated as: (100%-survival rate %). For
the concentration of cisplatin necessary to result in 50%
inhibition (IC50), the data was calculated using the
weighted linear regression method with SPSS 22.0 software (IBM
SPSS, Armonk, NY, USA). Each experiment was repeated >3
times.
Western blotting
Cells were treated with cisplatin (20 µM),
CHIR-99021 (3 µM), XAV-939 (1 µM), cisplatin + CHIR-99021 (20 and 3
µM, respectively), cisplatin + XAV-939 (20 and 1 µM, respectively)
or RPMI-1640 alone for 48 h, as previously described. Then, total
protein was extracted by treatment with RIPA lysis buffer (P0013B;
Beyotime Institute of Biotechnology) for 45 min on ice,
centrifugation (10,000 × g) for 25 min at 4°C and collection
of the supernatants, which were stored at −20°C until required.
Total protein concentration was determined using the BCA Protein
Assay Kit (P0010S; Beyotime Institute of Biotechnology). Equal
quantities of protein preparations (50 µg) were separated by 12%
SDS-PAGE and transferred onto polyvinylidene fluoride membranes.
The membranes were then blocked with 5% non-fat milk at room
temperature for between 1 and 2 h and incubated overnight with
primary antibodies against the following proteins: β-catenin
(1:1,000), JNK (1:2,000), CaMKII (1:1,000) and β-actin (1:5,000).
Next, membranes were incubated with the appropriate
fluorescently-labeled secondary antibodies (1:10,000). Between each
step, protein bands were washed with 1X PBST. Protein bands were
detected using an Odyssey Infrared Imaging System (Gene Company,
Ltd., Hong Kong, China). Quantitative analysis was performed using
ImageJ analysis software (National Institutes of Health, USA) with
β-actin serving as a control.
Immunofluorescence staining
Cells were cultured at 2×104 cells/well
into 24-well plates for 48 h and treated with cisplatin,
CHIR-99021, XAV-939, cisplatin + CHIR-99021, cisplatin + XAV-939 or
RPMI-1640 alone for 48 h, as previously described. Cells were then
fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton
X-100 and blocked with 5% horse serum (Vector Laboratories, Inc.,
Burlingame, CA, USA) in PBS-Tween for 30 min. Preparations were
incubated with primary antibodies [β-catenin (1:200), JNK (1:200)
and CaMKII (1:400)] as previously described at 4°C overnight and
then with the appropriate fluorescently-labeled secondary
antibodies (previously described; 1:1,000) for 60 min at room
temperature. In addition, nuclei were stained with DAPI. Between
each step, protein bands were washed with 1X PBST. Preparations
were visualized and images acquired using the EVOS XL Cell Imaging
System (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Statistical analysis
Data are presented as means ± the standard
deviation. Data was analyzed using SPSS software (version 22.0).
Differences between groups were statistically analyzed using the
Student's t-test or a paired t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Cisplatin sensitivity in SKOV3 and
SKOV3/DDP cell lines
To determine the sensitivity of SKOV3 and SKOV3/DDP
cells to cisplatin, cells were exposed to a gradient of cisplatin
concentrations (0–80 µM) for 24, 48 or 72 h and cell viability was
measured using the CCK-8 kit. The inhibition rate of SKOV3 and
SKOV3/DDP cells with cisplatin was found to increase in a
time-dependent and dose-dependent manner (Fig. 2A-C). There was a significant
difference between the concentrations of cisplatin required for
IC50 after 48 h in SKOV3 and SKOV3/DDP cells (6.086±0.38
vs. 26.135±1.825 µM, respectively; P<0.05; Fig. 2D), highlighting that SKOV3/DDP cells
were significantly cisplatin-resistant compared with SKOV3 cells.
From the analysis of these results, a dose of 20 µM cisplatin and
an exposure time of 48 h were used in subsequent studies.
Expression of β-catenin, JNK and
CaMKII in SKOV3 and SKOV3/DDP cell lines
To determine if cisplatin chemoresistance seen in
ovarian cancer cells was associated with deregulation of Wnt
signaling pathways, the present study evaluated the protein
expression levels of β-catenin, JNK and CaMKII in SKOV3 and
SKOV3/DDP cell lines. Quantification of western blotting results
(Fig. 3A) identified that expression
levels of β-catenin and JNK were significantly higher in SKOV3/DDP
cells compared with SKOV3 cells (*P<0.01,
&P<0.05; Fig. 3B).
However, CaMKII protein expression levels were lower in SKOV3/DPP
cells compared with SKOV3 cells. Following incubation with 20 µM
cisplatin for 48 h, β-catenin expression was reduced in SKOV3 and
SKOV3/DDP cells (*P<0.01, *P<0.001; Fig. 3B). Similarly, JNK protein expression
levels were significantly decreased in SKOV3 and SKOV3/DDP cells
following treatment with 20 µM cisplatin
(&P<0.05, #P<0.001; Fig. 3B). In contrast, cisplatin treatment
significantly increased CaMKII protein expression levels in SKOV3
and SKOV3/DDP cells (#P<0.001, *P<0.01; Fig. 3B). The difference in CaMKII
expression between SKOV3 and SKOV3/DDP cells was significant
following cisplatin treatment (&P<0.05; Fig. 3B). Similar results were observed in
immunofluorescence stains (Fig.
3C-E). These results suggest that abnormal activation of
Wnt/β-catenin and Wnt/JNK signaling pathways, and the abnormal
inactivation of the Wnt/Ca2+ signaling pathway, may be
associated with cisplatin chemoresistance in SKOV3/DPP cells.
 | Figure 3.Expression of β-catenin, JNK and
CaMKII in SKOV3 and SKOV3/DDP cells with and without the presence
of cisplatin. (A) Western blot for β-catenin, JNK and CaMKII
proteins from cells treated with cisplatin (20 µM) for 48 h and
untreated cells. (B) Quantification of protein expression levels
from western blotting. Each protein was normalized to β-actin.
&P<0.05, *P<0.01, #P<0.001. In
addition, immunofluorescence staining for the expression and
localization of (C) β-catenin, (D) JNK and (E) CaMKII proteins with
and without cisplatin (20 µg) was performed. JNK, c-Jun N-terminal
kinase; CaMKII, Ca2+/calmodulin-dependent protein kinase
II; DAPI, 4′,6-diamidino-2-phenylindole. |
Association between Wnt/β-catenin,
Wnt/JNK and Wnt/Ca2+ signaling pathways and ovarian
cancer cisplatin-resistance
To confirm the association observed between the
deregulation of Wnt signaling pathways and cisplatin-resistance in
ovarian cancer cells, CHIR-99021 (glycogen synthase kinase 3β
inhibitor, GSK3β) or XAV-939 (tankyrase inhibitor) were used in
addition to cisplatin to specifically activate or inhibit the
Wnt/β-catenin signaling pathway, respectively. The effect of these
drugs on SKOV3/DPP cell proliferation was then examined using the
CCK-8 kit. Compared with the control group without any drugs, the
majority of doses of CHIR-99021 or XAV-939 could not induce
significant cell death (Fig. 4A and
B). However, 3 µM CHIR-99021 combined with cisplatin
significantly suppressed the cytotoxicity of cisplatin (P<0.05;
Fig. 4A) and 1 µM XAV-939 combined
with cisplatin significantly increased the cytotoxicity of
cisplatin (P<0.05; Fig. 4B).
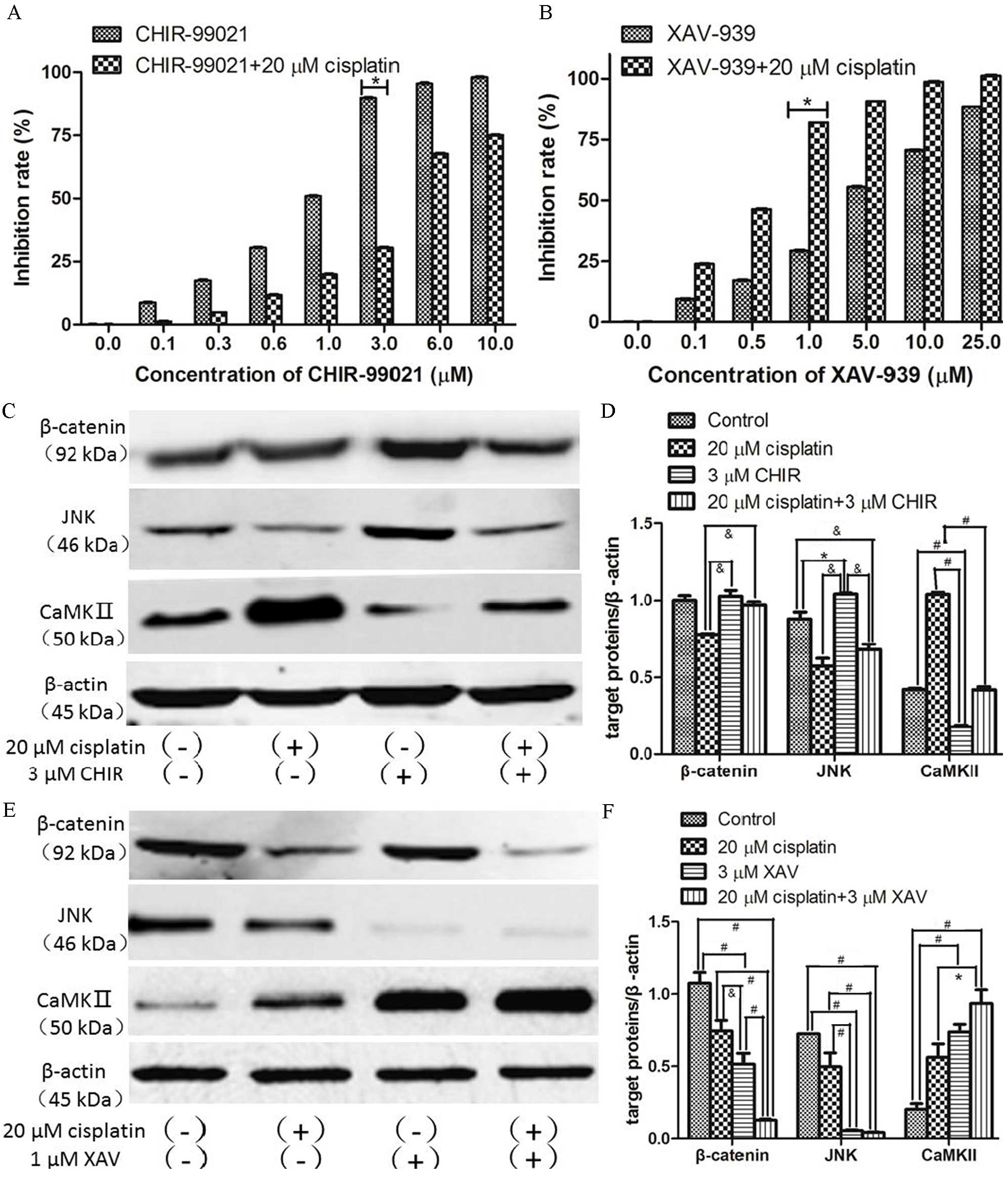 | Figure 4.Expression of β-catenin, JNK and
CaMKII in SKOV3/DDP cells following treatment with CHIR-99021 or
XAV-939 with and without cisplatin. SKOV3/DDP cells were treated
with a range of concentration of (A) CHIR-99021 or (B) XAV-939
alone or in combination with 20 µM cisplatin for 48 h. (C) Western
blot for β-catenin, JNK and CaMKII proteins from SKOV3/DDP cells
treated with CHIR-99021 (3 µM), cisplatin (20 µM) or both for 48 h.
(D) Quantification of protein expression levels from previous
western blot. Each protein was normalized to β-actin. (E) Western
blot for β-catenin, JNK and CaMKII proteins from SKOV3/DDP cells
treated with XAV-939 (1 µM), cisplatin (20 µM) or both for 48 h.
(F) Quantification of protein expression levels from previous
western blot. Each protein was normalized to β-actin.
&P<0.05, *P<0.01, #P<0.001. JNK,
c-Jun N-terminal kinase; CaMKII,
Ca2+/calmodulin-dependent protein kinase II; CHIR,
CHIR-99021; XAV, XAV-939; DAPI, 4′,6-diamidino-2-phenylindole. |
Next, SKOV3/DDP cells were treated with or without
cisplatin in the presence or absence of 3 µM CHIR-99021 or 1 µM
XAV-939, and the expression levels of β-catenin, JNK and CaMKII
were examined by western blotting and immunofluorescence staining.
Compared with the untreated group, the level of β-catenin was
increased by CHIR-99021 treatment (Fig.
4C and D). Futhermore, CHIR-99021 significantly reversed the
loss of β-catenin seen in cisplatin-treated cells
(&P<0.05; Fig.
4D), reduced cisplatin-induced loss of JNK
(&P<0.05) and significantly reduced
cisplatin-induced upregulation of CaMKII (#P<0.001;
Fig. 4D). In contrast, XAV-939
significantly decreased expression levels of β-catenin and JNK
(#P<0.001), and significantly increased expression
levels of CaMKII in cells (*P<0.01), with and without
co-treatment of cisplatin (Fig. 4E and
F). Interestingly, the expression levels of β-catenin and JNK
were positively correlated, and the level of CaMKII was negatively
correlated with β-catenin and JNK expression levels (Fig. 4E and F). Similar effects of
CHIR-99021 and XAV-939 were also observed in immunofluorescence
stains (Fig. 5). These results
indicate that the Wnt/β-catenin signaling pathway activation
antagonizes cisplatin-induced cell death, while the Wnt/β-catenin
signaling pathway inhibition enhances the cytotoxic effect of
cisplatin on ovarian cancer cells.
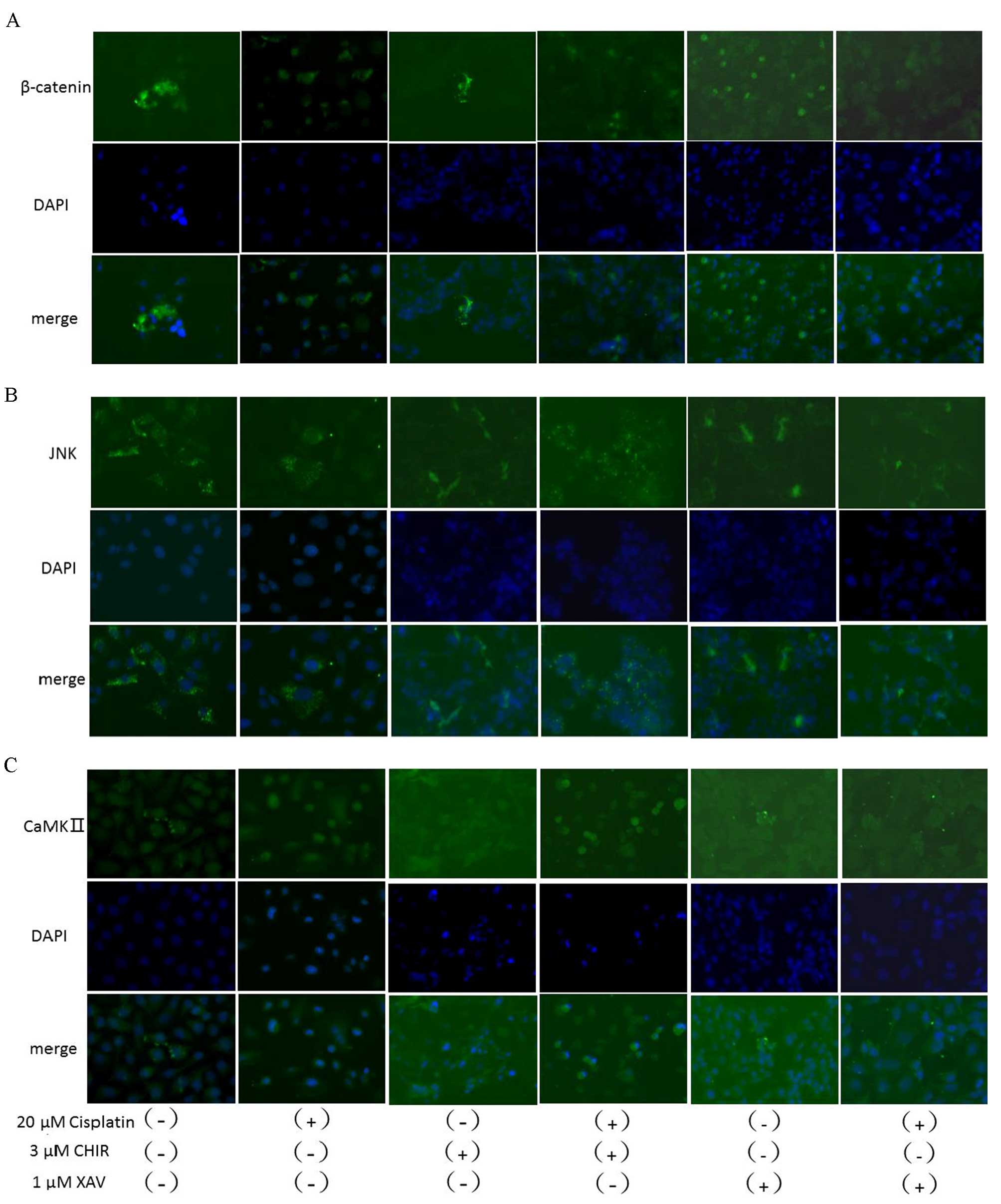 | Figure 5.Immunofluorescence staining for the
expression and localization of β-catenin, JNK and CaMKII treated
with and without cisplatin, CHIR-99021, XAV-939 or a combination.
SKOV3/DDP cells were immunofluorescence stained for (A) β-catenin,
(B) JNK or (C) CaMKII following treatment with cisplatin (20 µM),
CHIR-99021 (3 µM), XAV-939 (1 µM) or a combination for 48 h. JNK,
c-Jun N-terminal kinase; CaMKII,
Ca2+/calmodulin-dependent protein kinase II; CHIR,
CHIR-99021; XAV, XAV-939; DAPI, 4′,6-diamidino-2-phenylindole. |
Discussion
Ovarian cancer is the leading cause of mortality
among gynecological cancers (1). In
addition to cytoreductive surgery, cisplatin-based chemotherapy is
one of the most important therapeutic strategies for ovarian cancer
(2). Previous studies have shown
that Wnt signaling pathway deregulation correlates with ovarian
cancer cell proliferation, survival, invasion and metastasis
(25,26,28,30,31).
Furthermore, cisplatin chemoresistance has been an obstacle in the
successful treatment of ovarian cancer (8,32,33).
Several studies have concluded that aberrant regulation of Wnt
signaling induces tumor cell chemoresistance (16–18).
However, the molecular mechanisms by which Wnt signaling leads to
cisplatin chemoresistance remain poorly understood.
In the present study, a specific Wnt/β-catenin
signaling pathway activator (CHIR-99021) and inhibitor (XAV-939)
were used to explore the association between the deregulation of
Wnt signaling and cisplatin-resistance in ovarian cancer cells.
Firstly, the human ovarian cancer adenocarcinoma cell line SKOV3
and its cisplatin-resistant variant, SKOV3/DDP, were selected as
cell models. The IC50 value of cisplatin in SKOV3/DDP
cells was approximately four-fold that of SKOV3 cells, showing that
SKOV3/DDP cells were more cisplatin-resistant. Thus, these cell
lines are suitable models for the exploring cisplatin
chemoresistance.
Previous studies of the endometrioid subtype of
ovarian cancer have found mutations of CTNNB1 and abnormal
expression of β-catenin (22,23). The
present study demonstrated that β-catenin and JNK expression levels
were increased in cisplatin-resistant compared with
cisplatin-sensitive ovarian cancer cells. In contrast, CaMKII
expression levels were lower in cisplatin-resistant ovarian cancer
cells.
The correlation of the Wnt/β-catenin and
non-canonical Wnt signaling pathways in cisplatin chemoresistance
is controversial. Some investigators believe that both canoncial
and noncanoncial Wnt signaling pathways may promote each other in
cisplatin chemoresistance (34).
However, other research suggests that the Wnt/β-catenin signaling
pathway is crucial to cisplatin chemoresistance and that the
non-canonical Wnt signaling pathways obstruct this action (35–37). In
the current study, the Wnt/β-catenin signaling pathway agonist
CHIR-99021, which specifically inhibits GSK3β, was used to
investigate the correlation between the Wnt/β-catenin and
non-canonical Wnt signaling pathways. The SKOV3/DPP cell inhibition
rate with CHIR-99021 in combination with cisplatin was lower
compared with that of cisplatin alone. The level of β-catenin was
increased as a result of CHIR-99021 treatment, due to Wnt/β-catenin
signaling pathway activation. In addition, CHIR-99021 in
combination with cisplatin enhanced expression of JNK and
significantly decreased the expression of CaMKII compared with
cisplatin alone. Similarly, Zhao et al (21) found an association between high
expression levels of β-catenin and cisplatin chemoresistance in
A2780/DDP cells. This suggests that interference with β-catenin
expression could partly reverse cisplatin chemoresistance in
ovarian cancer cells.
SKOV3/DDP cells co-treated with XAV-939, a selective
Wnt/β-catenin signaling pathway inhibitor, and cisplatin produced a
greater inhibition of proliferation than when applied alone.
Expression of β-catenin and JNK was significantly reduced, and
expression of CaMKII was significantly higher, in SKOV3/DPP cells
following co-treatment with XAV-939 and cisplatin compared with
untreated cells or cells treated with cisplatin alone. Furthermore,
the expression levels of β-catenin and JNK were positively
correlated, while the level of CaMKII was negatively correlated
with of β-catenin and JNK expression levels.
In conclusion, the results of the present study
identify that, in SKOV3/DDP cells, the Wnt/β-catenin and Wnt/JNK
signaling pathways are positively correlated with cisplatin
chemoresistance, while the Wnt/Ca2+ signaling pathway is
negatively correlated with cisplatin chemoresistance. This suggests
that inhibiting the Wnt/β-catenin and Wnt/JNK signaling pathways,
and activating the Wnt/Ca2+ signaling pathway, could
reverse cisplatin-resistance in ovarian cancer cells. Developing
specific inhibitors and activators for these signaling pathways may
provide a treatment for cisplatin-resistant ovarian cancer, and the
key players of canonical (β-catenin) and non-canonical (JNK and
CaMKII) Wnt signaling are potential targets for drug
development.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bristow R, Chang J, Ziogas A, et al: NCCN
treatment guidelines for ovarian cancer: A population-based
vali-dation study of structural and process quality measures.
Gynecologic Oncology. 130:e182013. View Article : Google Scholar
|
|
3
|
Seiya S and Hiroaki I: Ovarian cancer and
drug resistance. Current Obstetrics and Gynecology Reports.
4:18–25. 2015. View Article : Google Scholar
|
|
4
|
Lloyd KL, Cree IA and Savage RS:
Prediction of resistance to chemotherapy in ovarian cancer: A
systematic review. BMC Cancer. 15:1172015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leamon PC, Lovejoy CD and Nguyen B:
Patient selection and targeted treatment in the management of
platinum-resistant ovarian cancer. Pharmgenomics Pers Med.
6:113–125. 2013.PubMed/NCBI
|
|
6
|
Go RS and Adjei AA: Review of the
comparative pharmacology and clinical activity of cisplatin and
carboplatin. J Clin Oncol. 17:409–422. 1999.PubMed/NCBI
|
|
7
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Claudia M, Jonathan AL and Pierluigi BP:
An overview of early investigational therapies for chemoresistant
ovarian cancer. Expert Opin Investig Drugs. 24:1163–1183. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sorrentino A, Liu CG, Addario A, Peschle
C, Scambia G and Ferlini C: Role of microRNAs in drug-resistant
ovarian cancer cells. Gynecol Oncol. 111:478–486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bayerlová M, Klemm F, Kramer F, Pukrop T,
Beißbarth T and Bleckmann A: Newly constructed network models of
different WNT signaling cascades applied to breast cancer
expression data. PLoS One. 10:e01440142015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shin H, Kim JH, Lee YS and Lee YC: Change
in gene expression profiles of secreted frizzled-related proteins
(SFRPs) by sodium butyrate in gastric cancers: Induction of
promoter demethylation and histone modification causing inhibition
of Wnt signaling. Int J Oncol. 40:1533–1542. 2012.PubMed/NCBI
|
|
13
|
Xi Y and Chen Y: Wnt signaling pathway:
Implications for therapy in lung cancer and bone metastasis. Cancer
Lett. 353:8–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sidaway P: Prostate cancer: Wnt signaling
induces resistance. Nat Rev Urol. 12:5972015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dellinger T, Warden C, Han E, et al: Wnt
pathway gene expression and association with clinicopathologic
characteristics in endometrial cancer-An analysis of The Cancer
Genome Atlas (TCGA). Gynecologic Oncology. 130:e892013. View Article : Google Scholar
|
|
16
|
Rosanò L, Cianfrocca R, Tocci P, Spinella
F, Di Castro V, Caprara V, Semprucci E, Ferrandina G, Natali PG and
Bagnato A: Endothelin A receptor/β-arrestin signaling to the Wnt
pathway renders ovarian cancer cells resistant to chemotherapy.
Cancer Res. 74:7453–7464. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Su HY, Lai HC, Lin YW, Liu CY, Chen CK,
Chou YC, Lin SP, Lin WC, Lee HY and Yu MH: Epigenetic silencing of
SFRP5 is related to malignant phenotype and chemoresistance of
ovarian cancer through Wnt signaling pathway. Int J Cancer.
127:555–567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laura R, Roberta C, Piera T, et al:
Endothelin A receptor/β-arrestin signaling to the Wnt pathway
renders ovarian cancer cells resistant to chemotherapy. Cancer
Research. 74:7463–7464. 2014.
|
|
19
|
Dawson K, Aflaki M and Nattel S: Role of
the Wnt-Frizzled system in cardiac pathophysiology: A rapidly
developing, poorly understood area with enormous potential. J
Physiol. 591:1409–1432. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chiurillo MA: Role of the Wnt/β-catenin
pathway in gastric cancer: An in-depth literature review. World J
Exp Med. 5:84–102. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao H, Wei W, Sun Y, Gao J, Wang Q and
Zheng J: Interference with the expression of β-catenin reverses
cisplatin resistance in A2780/DDP cells and inhibits the
progression of ovarian cancer in mouse model. DNA Cell Biol.
34:55–62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barghout SH, Zepeda N, Xu Z, Steed H, Lee
CH and Fu Y: Elevated β-catenin activity contributes to carboplatin
resistance in A2780cp ovarian cancer cells. Biochem Biophys Res
Commun. 468:173–178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Arend RC, Londoño-Joshi AI, Straughn JM Jr
and Buchsbaum DJ: The Wnt/β-catenin pathway in ovarian cancer: A
review. Gynecol Oncol. 131:772–779. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Matsumoto T, Yamazaki M, Takahashi H,
Kajita S, Suzuki E, Tsuruta T and Saegusa M: Distinct β-Catenin and
PIK3CA mutation profiles in endometriosis-associated ovarian
endometrioid and clear cell carcinomas. Am J Clin Pathol.
144:452–463. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McConechy MK, Ding J, Senz J, Yang W,
Melnyk N, Tone AA, Prentice LM, Wiegand KC, McAlpine JN, Shah SP,
et al: Ovarian and endometrial endometrioid carcinomas have
distinct CTNNB1 and PTEN mutation profiles. Mod Pathol. 27:128–134.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Forda CE, Punnia-Moorthya G, Henrya CE,
Llamosas E, Nixdorf S, Olivier J, Caduff R, Ward RL and
Heinzelmann-Schwarz V: The non-canonical Wnt ligand, Wnt5a, is
upregulated and associated with epithelial to mesenchymal
transition in epithelial ovarian cancer. Gynecol Oncol.
134:338–345. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Niehrs C: The complex world of WNT
receptor signaling. Nat Rev Mol Cell Biol. 13:767–779. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cizelsky W, Tata A, Kühl M and Kühl SJ:
The Wnt/JNK signaling target gene alcam is required for embryonic
kidney development. Development. 141:2064–2074. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tong XG, Barbourb M, Houc K, Gao C, Cao S,
Zheng J, Zhao Y, Mu R and Jiang HR: Interleukin-33 predicts poor
prognosis and promotes ovarian cancer cell growth and metastasis
through regulating ERK and JNK signaling pathways. Mol Oncol.
10:113–125. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ye XL, Zhao YR, Weng GB, Chen YC, Wei XN,
Shao JP and Ji H: IL-33-induced JNK pathway activation confers
gastric cancer chemotherapy resistance. Oncol Rep. 33:2746–2752.
2015.PubMed/NCBI
|
|
31
|
De A: Wnt/Ca2+ signaling
pathway: A brief overview. Acta Biochim Biophys Sin (Shanghai).
43:745–756. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ali AY, Farrand L, Kim JY, Byun S, Suh JY,
Lee HJ and Tsang BK: Molecular determinants of ovarian cancer
chemoresistance: New insights into an old conundrum. Ann N Y Acad
Sci. 1271:58–67. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Poisson LM, Munkarah A, Madi H, Datta I,
Hensley-Alford S, Tebbe C, Buekers T, Giri S and Rattan R: A
metabolomic approach to identifying platinum resistance in ovarian
cancer. J Ovarian Res. 8:132015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Luna-Ulloa LB, Hernández-Maqueda JG,
Castañeda-Patlán MC and Robles-Flores M: Protein kinase C in Wnt
signaling: Implications in cancer initiation and progression. IUBMB
Life. 63:915–921. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kagermeier-Schenk B, Wehner D, Özhan-Kizil
G, Yamamoto H, Li J, Kirchner K, Hoffmann C, Stern P, Kikuchi A,
Schambony A and Weidinger G: Waif1/5T4 inhibits Wnt/β-catenin
signaling and activates noncanonical Wnt pathways by modifying LRP6
subcellular localization. Dev Cell. 21:1129–1143. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
King TD, Zhang W, Suto MJ and Li Y:
Frizzled7 as an emerging target for cancer therapy. Cell Signal.
24:846–851. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ishitani T, Kishida S, Hyodo-Miura J, Ueno
N, Yasuda J, Waterman M, Shibuya H, Moon RT, Ninomiya-Tsuji J and
Matsumoto K: The TAK1-NLK mitogen-activated protein kinase cascade
functions in the Wnt-5a/Ca(2+) pathway to antagonize
Wnt/beta-catenin signaling. Mol Cell Biol. 23:131–139. 2003.
View Article : Google Scholar : PubMed/NCBI
|















