Introduction
Dilated cardiomyopathy (DCM) is the most frequent
type of non-ischemic cardiomyopathy worldwide, with an estimated
prevalence of 1 in 2,500 people and an incidence of 7/100,000
people annually (1,2). DCM is characterized by dilatation and
reduced contractile function of the left and right ventricles,
which may lead to progressive heart failure and sudden
cardiac-associated mortality in 4.5–79.3% of sufferers at 3 years
(3). Therefore, the early diagnosis
and treatment of DCM is important in order to prevent a poor
prognosis.
The molecular mechanisms underlying DCM have been
extensively explored in an attempt to provide effective diagnostic
and therapeutic methods for DCM. These previous studies have
demonstrated that DCM is associated with mutations in genes
encoding cytoskeletal, contractile or inner-nuclear membrane
proteins (4,5), including actin, desmin (6) and lamins A and C (7). Furthermore, it has been demonstrated
that mutations in sarcomere protein genes account for ~10% of cases
of familial DCM (8,9). Mitochondrial DNA mutations and
mitochondrial abnormalities have also been implicated in the
pathogenesis of DCM by altering myocardial ATP generation (10), including mitochondrial Hsp40 which
has a crucial role in preventing DCM (11). As a result, various potential
biomarkers have been elucidated, including high-sensitivity cardiac
troponin T (12), serum matrix
metalloproteinase-3 (13) and serum
Tenascin-C (14). However, the
mechanisms and pathogenesis underlying DCM remain poorly
understood.
Microarray technology enables the measurement of
global gene expression levels, and thus facilitates the
identification of crucial genes and altered biological processes in
DCM (15–17). Although Barth et al (16) have previously identified a common
gene expression signature in DCM via microarray data, deeper
analysis of the massive gene expression dataset is required via
bioinformatics. Therefore, the present study reanalyzed gene
expression data (16) to identify
differentially expressed genes (DEGs) which may contribute to the
development of DCM. Following this, functional enrichment and
protein-protein interaction (PPI) analyses of the DEGs were
performed using various bioinformatics tools. These findings may
advance understanding of the molecular mechanisms underlying
DCM.
Materials and methods
Gene expression data
Gene expression dataset (accession number, GSE3585)
(16) was downloaded from Gene
Expression Omnibus (http://ncbi.nlm.nih.gov/geo/). The GSE3585 dataset
included seven heart biopsy samples obtained from patients with DCM
at time of transplantation (GSM82386-GSM82392) and five non-failing
(NF) heart biopsies from NF donor hearts (GSM82381-GSM82386). Gene
expression levels were measured using Affymetrix Human Genome U133A
Array (Affymetrix Inc., Santa Clara, CA, USA).
Data pre-processing and differential
analysis
Raw data (CEL file) were read by an Affy package
(http://bioconductor.org/packages/release/bioc/html/affy.html)
(18) of R (version 3.1.2;
http://r-project.org/) (19). Probes detected in >50% of samples
were retained. Background correction, data normalization and
determination of expression levels was conducted using a Robust
Multi-array Average analysis method (20).
Differential analysis was performed using the linear
model of the lmFit and empirical Bayes moderated t test provided by
the Limma package (http://bioconductor.org/packages/release/bioc/html/limma.html)
(21). |log Fold Change (FC)
|>0.3 and false discovery rate (FDR) <0.05 were set as the
cut-offs to screen DEGs between DCM and NF hearts.
Functional enrichment analysis
Functional enrichment analysis of the DEGs was
conducted using the Database for Annotation, Visualization and
Integration Discovery (version 6.7; http://david.abcc.ncifcrf.gov/) (22), with a two-tailed Fisher exact test
based on the hypergeometric distribution. Benjamini corrected
P-values of <0.05 were set as the cut-off to screen out
significant Gene Ontology biological process (GOBP) terms (23) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathways (24).
Construction of PPI network
A PPI network was constructed for the DEGs using the
Search Tool for the Retrieval of Interacting Genes (STRING; version
9.1; http://string-db.org/) (25). Interacting pairs with a combined
score>0.4 were selected for the construction of the PPI network.
Proteins serve as the ‘nodes’ in the network and each pairwise
protein interaction is represented by an undirected link. Degree
was calculated for each node, which corresponds to the number of
interactions of a protein with other proteins. Hub genes were
subsequently selected according to the degree.
Results
Microarray data analysis identified
DEGs between the DCM and control groups
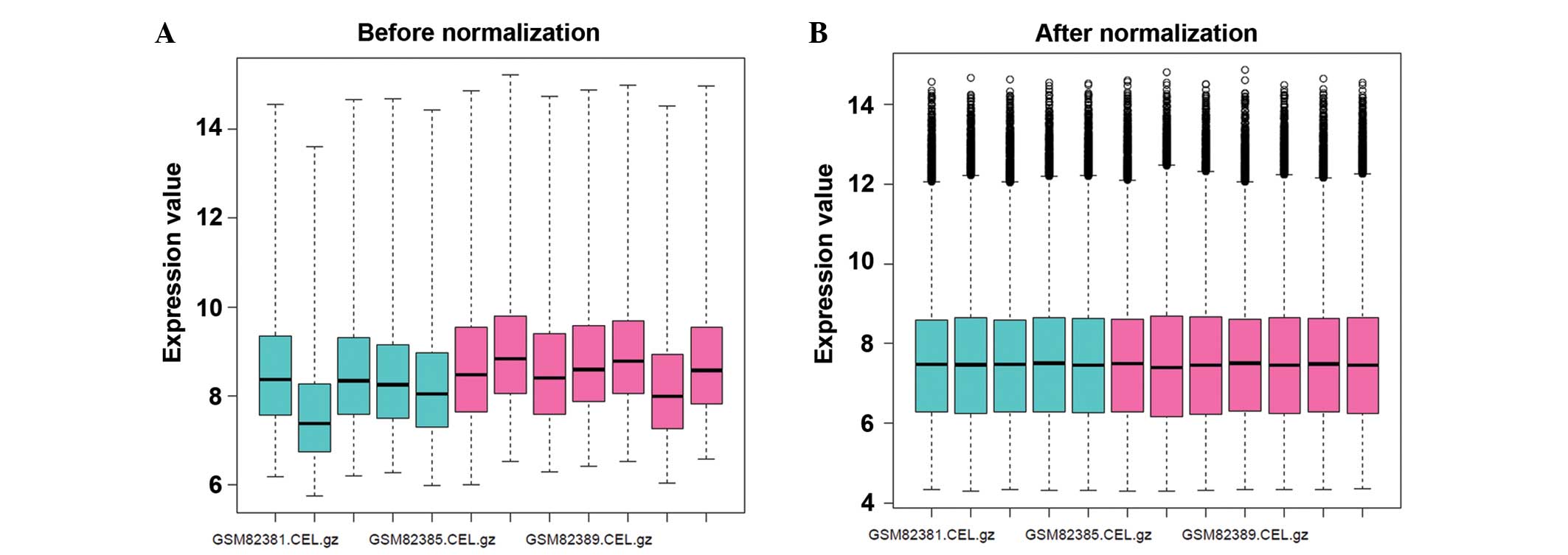
Following data normalization, the mean expression
levels of the gene expression profiles were presented in one
consistent line (Fig. 1).
Subsequently, according to the criteria of |log FC| >0.3 and FDR
<0.05, 89 DEGs were detected between the DCM and NF hearts,
including 22 downregulated and 67 upregulated genes. A greater
number of upregulated genes were detected, as compared with the
downregulated genes, which demonstrated that the DEGs had a
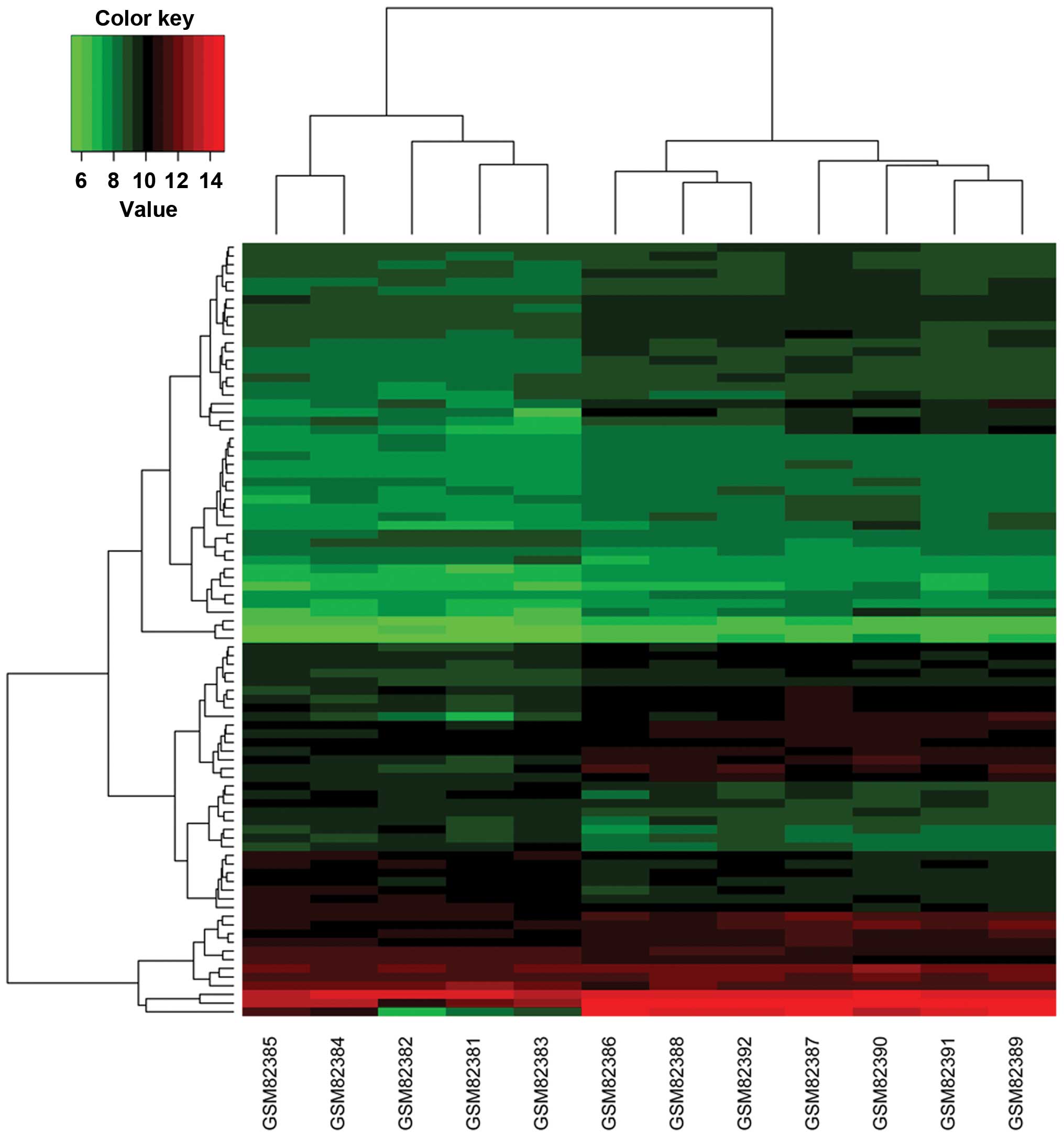
tendency to upregulate in DCM. Based on the DEGs, the DCM samples
were predominantly separated from the NF controls, implying the
reliability of the DEGs (Fig.
2).
GO and KEGG pathway analysis revealed
the functions of DEGs involved in DCM
Functional enrichment analysis was performed for the
downregulated and upregulated genes, respectively. For the
downregulated genes, two clusters were obtained using the Protein
Information Resource. The first cluster was demonstrated to be
associated with ‘chromosomal proteins’, including H1 histone family
member 0 (H1F0; logFC=−0.61; FDR=1.91E-02), H2A histone family
member Z (H2AFZ; logFC=−0.47; FDR=1.94E-02), histone cluster 1 H1c
(HIST1H1C; logFC=−0.71; FDR=3.69E-02) and histone cluster 1 H2bd
(HIST1H2BD; logFC=−0.69; FDR=2.89E-02). The second cluster was
associated with transmembrane transport-related proteins, including
STEAP family member 3 (logFC=−0.92; FDR=4.39E-02), potassium
voltage-gated channel subfamily D member 3 (logFC=−0.54;
FDR=4.48E-02) and insulin-like growth factor 2 receptor
(logFC=−0.44; FDR=4.76E-02). Furthermore, downregulated genes
(H1F0, HIST1H2BD, HIST1H1C and H2AFZ) were significantly enriched
in the biological processes of ‘nucleosome assembly’ (P=1.22E-04),
‘chromatin assembly’ (P=1.35E-04), ‘protein-DNA complex assembly’
(P=1.55E-04), ‘nucleosome organization’ (P=1.65E-04) and ‘DNA
packaging’ (P=3.25E-04; Table
I).
 | Table I.Significantly enriched functional
terms of downregulated genes. |
Table I.
Significantly enriched functional
terms of downregulated genes.
| Category | Enrichment
score | Terms | P-value | Genes |
|---|
|
SP_PIR_KEYWORDS |
|
|
|
|
Annotation Cluster 1 |
1.689911619479253 | Chromosomal
protein | 3.72E-04 | H1F0, HIST1H2BD,
HIST1H1C, H2AFZ |
|
Annotation Cluster 2 |
0.32418860902901725 | Transport | 2.25E-01 | STEAP3, KCND3,
IGF2R, SELENBP1 |
| GO_BP_FAT |
|
|
|
|
|
Annotation Cluster 1 |
2.7555946833424456 |
|
|
|
|
GO:0006334 |
| Nucleosome
assembly | 1.22E-04 | H1F0, HIST1H2BD,
HIST1H1C, H2AFZ |
|
GO:0031497 |
| Chromatin
assembly | 1.35E-04 | H1F0, HIST1H2BD,
HIST1H1C, H2AFZ |
|
GO:0065004 |
| Protein-DNA complex
assembly | 1.55E-04 | H1F0, HIST1H2BD,
HIST1H1C, H2AFZ |
|
GO:0034728 |
| Nucleosome
organization | 1.65E-04 | H1F0, HIST1H2BD,
HIST1H1C, H2AFZ |
|
GO:0006323 |
| DNA packaging | 3.25E-04 | H1F0, HIST1H2BD,
HIST1H1C, H2AFZ |
For the upregulated genes, two clusters were also
acquired using the Protein Information Resource, including
‘secreted proteins’ such as connective tissue growth factor (CTGF;
logFC=0.84; FDR=3.04E-02) and insulin-like growth factor binding
protein 3 (IGFBP3; logFC=0.84; FDR=3.04E-02) in cluster 1, and
‘phosphotransferase’ such as phosphatidylinositol-4,5-bisphosphate
3-kinase, catalytic subunit alpha (PIK3CA; logFC=0.59;
FDR=4.44E-02) and insulin receptor (INSR; logFC=0.61; FDR=2.27E-02)
in cluster 2. Additionally, six GOBP terms were detected in the
upregulated genes (P<0.05), including ‘cell adhesion’ (CTGF),
‘skeletal system development’ (CTGF and IGFBP3), ‘muscle organ
development’ (SMAD7; logFC=0.56; FDR=4.39E-02), and ‘regulation of
cell migration’ (SMAD7, IGFBP3 and INSR) (Table II). No significant pathways were
enriched for the DEGs.
| Table II.Significantly enriched functional
terms of upregulated genes. |
Table II.
Significantly enriched functional
terms of upregulated genes.
| Category | Enrichment
score | Terms | P-value | Genes |
|---|
|
SP_PIR_KEYWORDS |
|
|
|
|
Annotation cluster 1 |
1.1489102585231292 | Secreted | 9.05E-03 | AEBP1, LTBP1,
SPOCK1 FRZB, OMD, CTGF, CFH, NPPB, LOX, PRSS23, LAMB1, IGFBP3,
NPPA |
|
Annotation cluster 2 |
1.1141850825108304 |
Phosphotransferase | 2.94E-02 | ROR1, PIK3CA, CLK1,
INSR |
| GOTERM_BP_FAT |
|
|
|
|
|
Annotation cluster 1 |
2.187528735182074 |
|
|
|
|
GO:0007155 |
| Cell adhesion | 6.23E-03 | OMD, AEBP1, CTGF,
PKD2, SPOCK1, SGCE, DLG5, LAMB1, SSPN |
|
Annotation cluster 2 |
1.9708171852370016 |
|
|
|
|
GO:0001501 |
| Skeletal
development | 8.69E-03 | AEBP1, CTGF, EXT1,
FRZB, IGFBP3, CBFB |
|
Annotation cluster 3 |
1.5566908357862366 |
|
|
|
|
GO:0007517 |
| Muscle organ
development | 9.90E-03 | AEBP1, SMAD7, SGCE,
TPM1, MYH10 |
|
Annotation cluster 4 |
1.3188719375499292 |
|
|
|
|
GO:0030334 |
| Regulation of cell
migration | 4.57E-03 | SMAD7, LAMB1,
IGFBP3, INSR, TPM1 |
|
Annotation cluster 5 |
1.2806966535392923 |
|
|
|
|
GO:0007507 |
| Heart
development |
1.06E-02 | SMAD7, PKD2, INSR,
TPM1, MYH10 |
|
Annotation cluster 6 |
1.1685982822293663 |
|
|
|
|
GO:0006163 |
| Purine nucleotide
metabolic process | 3.80E-02 | MGEA5, ATP10D,
ATP13A3, NPPA |
PPI network analysis identifies
crucial genes for DCM
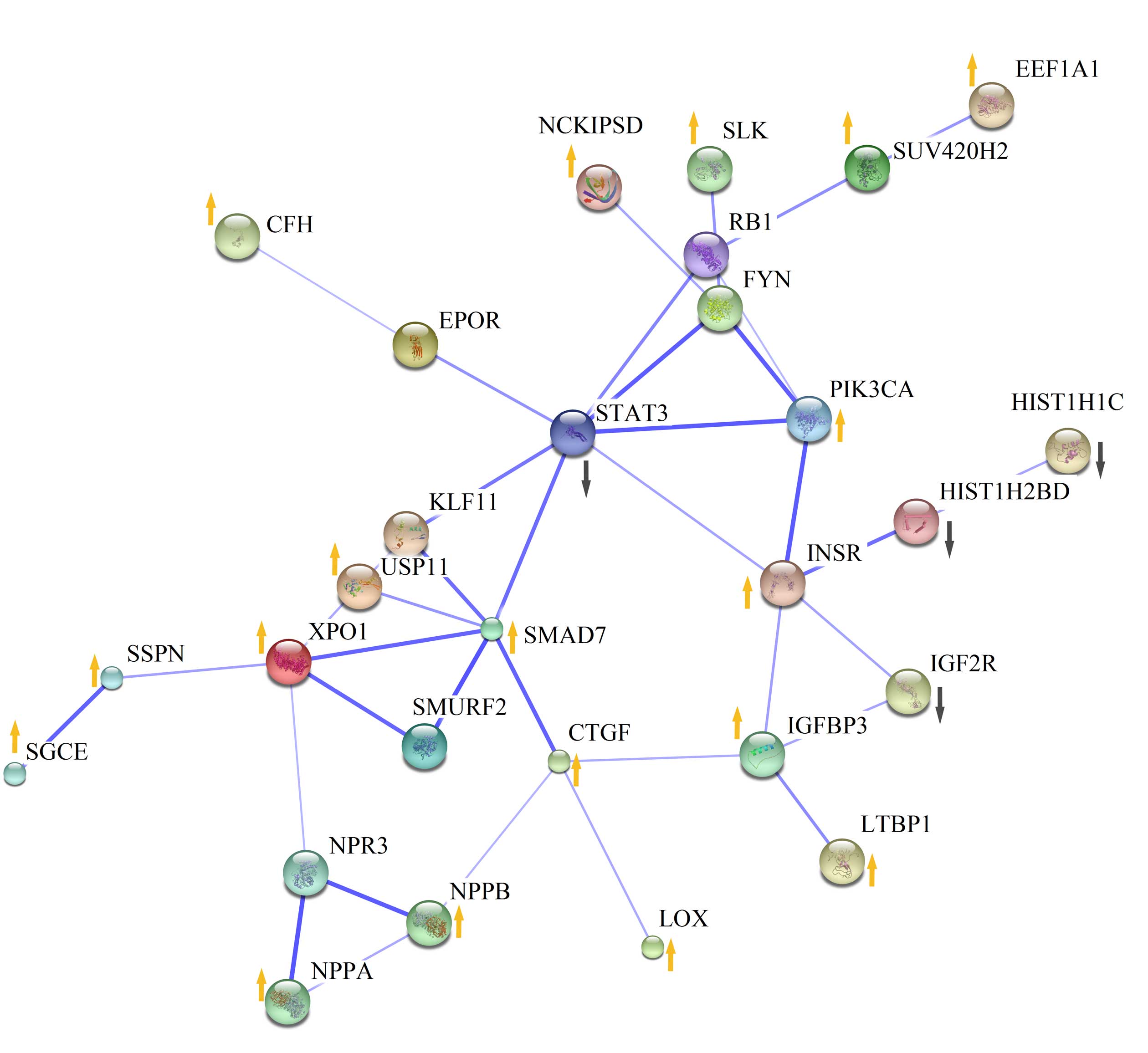
A PPI network containing 22 DEGs was constructed
using the protein interaction information obtained from STRING
(Fig. 3). By calculating the degree
of each gene in the network, seven DEGs were considered to be
crucial for DCM with a degree of >3, including signal transducer
and activator of transcription 3 (STAT3; degree, 7; logFC=−0.86;
FDR= 2.27E-02), SMAD7 (degree, 6), INSR (degree, 5), exportin 1
(XPO1; degree, 5; logFC=0.62; FDR=2.74E-02), CTGF (degree, 4),
IGFBP3 (degree, 4) and PIK3CA (degree, 4).
Discussion
In the present study, by reanalyzing the gene
expression profiles of DCM and NF samples (16), 89 DEGs were identified in DCM
patients, including 22 downregulated and 67 upregulated genes.
Downregulated genes were demonstrated to encode chromosomal
proteins and transmembrane transport-related proteins, which were
associated with ‘nucleosome assembly’, ‘chromatin assembly’,
‘protein-DNA complex assembly’, ‘nucleosome organization’ and ‘DNA
packaging’. Upregulated genes were enriched into two annotation
clusters, ‘secreted proteins’ and ‘phosphotransferase’, which were
associated with ‘cell adhesion’, ‘skeletal system development’,
‘muscle organ development’ and ‘regulation of cell migration’.
Notably, upregulated SMAD7, STAT3, INSR,
XPO1, CTGF, IGFBP3 and PIK3CA were hub
nodes in the PPI network.
Several histone family members were demonstrated to
be downregulated in DCM, including H1F0, H2AFZ,
HIST1H2BD and HIST1H1C. To the best of our knowledge,
this is the first report of the association between these genes and
DCM. Histones are basic nuclear proteins that are responsible for
the nucleosome structure of chromosomal fiber in eukaryotes
(26). Previous studies have
indicated that histone acetylation/deacetylation regulates cardiac
morphogenesis, growth and contractility (27,28). The
present results demonstrated that these downregulated histone
family member genes were significantly enriched in GO terms
including ‘nucleosome assembly’, ‘chromatin assembly’, ‘protein-DNA
complex assembly’, ‘nucleosome organization’ and ‘DNA packaging’.
Therefore, the downregulation of these genes may contribute to the
blockade of nucleosome formation. As previously described, the
untranscribed regions of eukaryotic genomes are packaged into
nucleosomes, which may repress gene expression in general (29). Depletion of nucleosomal histones may
lead to the silencing of specific genes (30). Therefore, we hypothesize that the
downregulation of histones may induce the overexpression of
numerous genes associated with the development of DCM via
nucleosome formation, which may explain the tendency for
upregulation of DEGs observed in the present study.
In the present study, 67 upregulated genes were
detected in DCM. CTGF, which is a profibrotic cytokine, was
demonstrated to be associated with ‘cell adhesion’ and ‘skeletal
system development’. Previous studies have indicated that CTGF has
a key role in the deleterious process of cardiac fibrosis, which is
a hallmark of DCM (31,32). Furthermore, it has been demonstrated
that CTGF is highly induced in viral myocarditis and its silencing
may counteract cardiac fibrosis and heart muscle dysfunction
(33). CTGF was also demonstrated to
be consistently upregulated in DCM patients in the present study,
which may contribute to abnormal cardiac fibrosis. Moreover, it has
been demonstrated that IGFBP3, which is a member of the
insulin-like growth factor binding protein family, is upregulated
in the failing hearts of DCM patients (34,35).
Hassfeld et al (36) regard
IGFBP3 as an independent predictors of a poor prognosis in patients
with DCM. Notably, the results of the present study indicated that
upregulated IGFBP3 was related to ‘skeletal system development’ and
‘regulation of cell migration’. Similarly, INSR was upregulated in
DCM and was associated with ‘regulation of cell migration’. The
binding of insulin to INSR stimulates glucose uptake which affects
muscle function in vitro (37), thus the upregulated INSR may be
associated with muscle contractility. Furthermore, SMAD7 was
upregulated in DCM patients, which is one feature of dysregulated
extracellular matrix degradation which may lead to cardiac fibrosis
in patients with diabetic cardiomyopathy (38). DCM is characterized by abnormal
contractile function (39,40). The DEGs mentioned above (CTGF, SMAD7,
IGFBP3 and INSR) were found to be enriched in ‘cell adhesion’,
‘skeletal system development’, ‘muscle organ development’ and
‘regulation of cell migration’, and abnormalities in these
processes may contribute to weakened contractile function. Notably,
CTGF, SMAD7, IGFBP3 and INSR were also demonstrated to be hub nodes
in the PPI network, suggesting their predominant roles among the
DEGs. These four DEGs may be future therapeutic targets for the
treatment of DCM.
Although the STAT3, XPO1 and PIK3CA GO terms were
not enriched in the present study, they did form hub nodes in the
PPI network, thus suggesting that these genes are also important in
DCM. This is supported by previous studies. Podewski et al
(41) observed that the protein
expression levels of STAT3 were significantly decreased in the
cardiomyocytes of patients with end-stage DCM. Furthermore, using
the knockout technique, Hilfiker-Kleiner et al (42) demonstrated reduced myocardial
capillary density and increased interstitial fibrosis in
STAT3-deficient mice, leading to DCM with impaired cardiac function
and premature mortality. PIK3CA, a stress-associated lipid kinase,
has been shown to be important for maintaining cardiac structure
and function, and its upregulation ultimately prolonged the
survival of a mouse model of DCM (43,44). In
addition, XPO1 has been reported to be a key nucleocytoplasmic
transport-related gene in DCM (45,46), and
upregulation of XPO1 may be involved in DCM by mediating the
nuclear export of several molecules that regulate cardiac growth
and development, including a member of the Rho family of GTPases,
Ras homolog family member U (RhoU), and a transcriptional
coactivator and cytoskeleton regulator, four and a half LIM domains
3 (FHL3) (46). It has been reported
that the loss of RhoU results in the mis-localization of cell
adhesion molecules, including Alcama and N-cadherin, to the
cytoplasm and the apical and basal cell membrane, instead of the
atrioventricular cardiomyocyte cell junctions, resulting in failure
to form the atrioventricular canal and loop the linear heart tube
and thus influencing cardiac function (47). Furthermore, downregulated FHL3
expression has been associated with the systolic dysfunction of DCM
patients by inhibiting the expression of myosin heavy chain isoform
2a (48,49).
In conclusion, the results of the present study
suggested that downregulated histones may lead to the
overexpression of DCM-specific genes via damage to the nucleosome.
Furthermore, upregulated genes, including CTGF, SMAD7, IGFBP3 and
INSR, may lead to weakened contractile function via various
biological pathways associated with muscle development. STAT3, XPO1
and PIK3CA may also be important for maintaining heart function.
There were some limitations to the results of the present study.
The DEGs identified by microarray data were not validated using
real-time polymerase chain reaction and there was no intervention
or patient follow-up performed. Despite these limitations, the
present findings may advance the understanding of the pathogenesis
of DCM and provide novel targets for clinical treatment and
diagnosis.
Acknowledgements
The present study was supported by the Natural
Science Foundation of China (grant no. 31360227).
References
|
1
|
Hershberger RE, Hedges DJ and Morales A:
Dilated cardiomyopathy: The complexity of a diverse genetic
architecture. Nat Rev Cardiol. 10:531–547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jefferies JL and Towbin JA: Dilated
cardiomyopathy. Lancet. 375:752–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goldberger JJ, SubaÄ H, Patel T, Cunnane R
and Kadish AH: Sudden cardiac death risk stratification in patients
with nonischemic dilated cardiomyopathy. J Am Coll Cardiol.
63:1879–1889. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gerull B, Gramlich M, Atherton J, McNabb
M, Trombitás K, Sasse-Klaassen S, Seidman JG, Seidman C, Granzier
H, Labeit S, et al: Mutations of TTN, encoding the giant muscle
filament titin, cause familial dilated cardiomyopathy. Nat Genet.
30:201–204. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Villard E, Duboscq-Bidot L, Charron P,
Benaiche A, Conraads V, Sylvius N and Komajda M: Mutation screening
in dilated cardiomyopathy: Prominent role of the beta myosin heavy
chain gene. Eur Heart J. 26:794–803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Taylor MR, Slavov D, Ku L, Di Lenarda A,
Sinagra G, Carniel E, Haubold K, Boucek MM, Ferguson D, Graw SL, et
al: Prevalence of desmin mutations in dilated cardiomyopathy.
Circulation. 115:1244–1251. 2007.PubMed/NCBI
|
|
7
|
Schmidt HH and Lochs H: Lamin A/C gene
mutation associated with dilated cardiomyopathy with variable
skeletal muscle involvement. Circulation. 103:E202001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frazier AH, Ramirez-Correa GA and Murphy
AM: Molecular mechanisms of sarcomere dysfunction in dilated and
hypertrophic cardiomyopathy. Prog Pediatr Cardiol. 31:29–33. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kamisago M, Sharma SD, DePalma SR, Solomon
S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED,
et al: Mutations in sarcomere protein genes as a cause of dilated
cardiomyopathy. N Engl J Med. 343:1688–1696. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arbustini E, Diegoli M, Fasani R, Grasso
M, Morbini P, Banchieri N, Bellini O, Dal Bello B, Pilotto A,
Magrini G, et al: Mitochondrial DNA mutations and mitochondrial
abnormalities in dilated cardiomyopathy. Am J Pathol.
153:1501–1510. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hayashi M, Imanaka-Yoshida K, Yoshida T,
Wood M, Fearns C, Tatake RJ and Lee JD: A crucial role of
mitochondrial Hsp40 in preventing dilated cardiomyopathy. Nat Med.
12:128–132. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kawahara C, Tsutamoto T, Nishiyama K,
Yamaji M, Sakai H, Fujii M, Yamamoto T and Horie M: Prognostic role
of high-sensitivity cardiac troponin T in patients with nonischemic
dilated cardiomyopathy. Circ J. 75:656–661. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ohtsuka T, Nishimura K, Kurata A, Ogimoto
A, Okayama H and Higaki J: Serum matrix metalloproteinase-3 as a
novel marker for risk stratification of patients with nonischemic
dilated cardiomyopathy. J Card Fail. 13:752–758. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Terasaki F, Okamoto H, Onishi K, Sato A,
Shimomura H, Tsukada B, Imanaka-Yoshida K, Hiroe M, Yoshida T,
Kitaura Y, et al: Higher serum tenascin-C levels reflect the
severity of heart failure, left ventricular dysfunction and
remodeling in patients with dilated cardiomyopathy. Circ J.
71:327–330. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barrans JD, Allen PD, Stamatiou D, Dzau VJ
and Liew CC: Global gene expression profiling of end-stage dilated
cardiomyopathy using a human cardiovascular-based cDNA microarray.
Am J Pathol. 160:2035–2043. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barth AS, Kuner R, Buness A, Ruschhaupt M,
Merk S, Zwermann L, Kääb S, Kreuzer E, Steinbeck G, Mansmann U, et
al: Identification of a common gene expression signature in dilated
cardiomyopathy across independent microarray studies. J Am Coll
Cardiol. 48:1610–1617. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Camargo A and Azuaje F: Identification of
dilated cardiomyopathy signature genes through gene expression and
network data integration. Genomics. 92:404–413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gentleman RC, Carey VJ, Bates DM, Bolstad
B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al:
Bioconductor: Open software development for computational biology
and bioinformatics. Genome Biol. 5:R802004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Smyth GK: Limma: Linear models for
microarray data. Journal. 397–420. 2005.
|
|
22
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology. Nat Genet. 25:25–29. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9.1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res.
41:(Database Issue). D808–D815. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thakar A, Gupta P, Ishibashi T, Finn R,
Silva-Moreno B, Uchiyama S, Fukui K, Tomschik M, Ausio J and
Zlatanova J: H2A.Z and H3.3 histone variants affect nucleosome
structure: Biochemical and biophysical studies. Biochemistry.
48:10852–10857. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Backs J and Olson EN: Control of cardiac
growth by histone acetylation/deacetylation. Circ Res. 98:15–24.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Montgomery RL, Davis CA, Potthoff MJ,
Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA and Olson EN:
Histone deacetylases 1 and 2 redundantly regulate cardiac
morphogenesis, growth and contractility. Genes Dev. 21:1790–1802.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Orphanides G and Reinberg D: A unified
theory of gene expression. Cell. 108:439–451. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wyrick JJ, Holstege FC, Jennings EG,
Causton HC, Shore D, Grunstein M, Lander ES and Young RA:
Chromosomal landscape of nucleosome-dependent gene expression and
silencing in yeast. Nature. 402:418–421. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Touvron M, Escoubet B, Mericskay M,
Angelini A, Lamotte L, Santini MP, Rosenthal N, Daegelen D, Tuil D
and Decaux JF: Locally expressed IGF1 propeptide improves mouse
heart function in induced dilated cardiomyopathy by blocking
myocardial fibrosis and SRF-dependent CTGF induction. Dis Model
Mech. 5:481–491. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
van Almen GC, Verhesen W, van Leeuwen RE,
van de Vrie M, Eurlings C, Schellings MW, Swinnen M, Cleutjens JP,
van Zandvoort MA, Heymans S and Schroen B: MicroRNA-18 and
microRNA-19 regulate CTGF and TSP-1 expression in age-related heart
failure. Aging Cell. 10:769–779. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lang C, Sauter M, Szalay G, Racchi G,
Grassi G, Rainaldi G, Mercatanti A, Lang F, Kandolf R and Klingel
K: Connective tissue growth factor: A crucial cytokine-mediating
cardiac fibrosis in ongoing enterovirus myocarditis. J Mol Med
(Berl). 86:49–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Broglio F, Benso A, Arvat E, Aimaretti G,
Gottero C, Granata R, Boghen MF, Bobbio M, Camanni F and Ghigo E:
Normal IGF-I and enhanced IGFBP-3 response to very low rhGH dose in
patients with dilated cardiomyopathy. J Endocrinol Invest.
23:520–525. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pucci A, Zanini C, Granata R, Ghignone R,
Iavarone A, Broglio F, Sorrentino P, Bergamasco L, Rinaldi M and
Ghigo E: Myocardial insulin-like growth factor-1 and insulin-like
growth factor binding protein-3 gene expression in failing hearts
harvested from patients undergoing cardiac transplantation. J Heart
Lung Transplant. 28:402–405. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hassfeld S, Eichhorn C, Stehr K, Naegele
H, Geier C, Steeg M, Ranke MB, Oezcelik C and Osterziel KJ:
Insulin-like growth factor-binding proteins 2 and 3 are independent
predictors of a poor prognosis in patients with dilated
cardiomyopathy. Heart. 93:359–360. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Khodabukus A and Baar K: Glucose
concentration and streptomycin alter in vitro muscle function and
metabolism. J Cell Physiol. 230:1226–1234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Van Linthout S, Seeland U, Riad A,
Eckhardt O, Hohl M, Dhayat N, Richter U, Fischer JW, Böhm M,
Pauschinger M, et al: Reduced MMP-2 activity contributes to cardiac
fibrosis in experimental diabetic cardiomyopathy. Basic Res
Cardiol. 103:319–327. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lakdawala NK, Thune JJ, Colan SD, Cirino
AL, Farrohi F, Rivero J, McDonough B, Sparks E, Orav EJ, Seidman
JG, et al: Subtle abnormalities in contractile function are an
early manifestation of sarcomere mutations in dilated
cardiomyopathy. Circ Cardiovasc Genet. 5:503–510. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Parsons JT, Horwitz AR and Schwartz MA:
Cell adhesion: Integrating cytoskeletal dynamics and cellular
tension. Nat Rev Mol Cell Biol. 11:633–643. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Podewski EK, Hilfiker-Kleiner D, Hilfiker
A, Morawietz H, Lichtenberg A, Wollert KC and Drexler H:
Alterations in Janus kinase (JAK)-signal transducers and activators
of transcription (STAT) signaling in patients with end-stage
dilated cardiomyopathy. Circulation. 107:798–802. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hilfiker-Kleiner D, Hilfiker A, Fuchs M,
Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z,
Podewski E, et al: Signal transducer and activator of transcription
3 is required for myocardial capillary growth, control of
interstitial matrix deposition, and heart protection from ischemic
injury. Circ Res. 95:187–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
McMullen JR, Amirahmadi F, Woodcock EA,
Schinke-Braun M, Bouwman RD, Hewitt KA, Mollica JP, Zhang L, Zhang
Y, Shioi T, et al: Protective effects of exercise and
phosphoinositide 3-kinase (p110α) signaling in dilated and
hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 104:612–617.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Waardenberg AJ, Bernardo BC, Ng DC,
Shepherd PR, Cemerlang N, Sbroggiò M, Wells CA, Dalrymple BP,
Brancaccio M, Lin RC and McMullen JR: Phosphoinositide 3-kinase
(PI3K (p110α)) directly regulates key components of the Z-disc and
cardiac structure. J Biol Chem. 286:30837–30846. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Molina-Navarro MM, Roselló-Lletí E,
Tarazón E, Ortega A, Sánchez-Izquierdo D, Lago F, González-Juanatey
JR, García-Pavía P, Salvador A, Montero JA, et al: Heart failure
entails significant changes in human nucleocytoplasmic transport
gene expression. Int J Cardiol. 168:2837–2843. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Molina-Navarro MM, Triviño JC,
Martínez-Dolz L, Lago F, González-Juanatey JR, Portolés M and
Rivera M: Functional networks of nucleocytoplasmic
transport-related genes differentiate ischemic and dilated
cardiomyopathies. A new therapeutic opportunity. PLoS One.
9:e1047092014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dickover M, Hegarty JM, Ly K, Lopez D,
Yang H, Zhang R, Tedeschi N, Hsiai TK and Chi NC: The atypical Rho
GTPase, RhoU, regulates cell-adhesion molecules during cardiac
morphogenesis. Dev Biol. 389:182–191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Y, Li W, Zhu M, Li Y, Xu Z and Zuo
B: FHL3 differentially regulates the expression of MyHC isoforms
through interactions with MyoD and pCREB. Cell Signal. 28:60–73.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Abraham WT, Gilbert EM, Lowes BD, Minobe
WA, Larrabee P, Roden RL, Dutcher D, Sederberg J, Lindenfeld JA,
Wolfel EE, et al: Coordinate changes in Myosin heavy chain isoform
gene expression are selectively associated with alterations in
dilated cardiomyopathy phenotype. Mol Med. 8:750–760.
2002.PubMed/NCBI
|