Introduction
The thyrotropin receptor (TSHR) is a
G-protein-coupled receptor consisting of seven
transmembrane-spanning regions and a large extracellular domain
that mediates the effect of TSH in thyroid development, growth and
hormone synthesis. TSH exerts its biological effects through
binding with TSHR to activate different effectors, predominantly
adenyl cyclase and phospholipase C (1). Previous molecular analyses have
revealed that mutations in the TSHR gene result in various thyroid
dysfunctions (2).
Subclinical hypothyroidism (SH) is a clinical
condition defined as an elevated serum TSH level associated with
normal free thyroxine (FT4) and free triiodothyronine (FT3). The
patient is usually asymptomatic; however, a thorough evaluation of
patients has indicated consequences for quality of life, cognitive
abilities, cholesterol metabolism, heart rate, bone mineral density
and atherogenesis (3). Chronic
autoimmune thyroiditis is the most frequent cause of subclinical
hypothyroidism in adults (4), and
its diagnostic hallmarks are circulating thyroglobulin antibody
(TgAb), thyroid peroxidase antibody (TPOAb) or TSHR-blocking
antibodies. A hypoechogenic pattern of the thyroid gland at
ultrasound examination is also observed in autoimmune thyroiditis
(5). In rare cases, resistance to
TSH is the cause of SH. Resistance to TSH is a syndrome in which
the thyroid displays a variable degree of hyposensitivity to a
biologically active TSH molecule (6). The condition may be a result of
abnormalities in the TSHR (7–13).
Loss-of-function (LOF) mutations of TSHR always cause
hypothyroidism, with the severity ranging from SH to overt
hypothyroidism depending on the degree of the mutation (14). The present study reported the case of
a woman with SH with a TSHR mutation (V87L) who tested negative for
circulating TgAb and TPOAb.
Patients and methods
Case report
A 59-year-old female patient with a 12-year history
of goiter was admitted to the Department of General Surgery, Xin
Hua Hospital affiliated to Shanghai Jiaotong University School of
Medicine in May 2010 for thyroid surgery. The patient was not
receiving any medication at the time and the primary complaint was
of pain in the neck region. The present study was approved by the
Ethics Committee of Xinhua Hospital Affiliated to Shanghai Jiaotong
University School of Medicine. Written informed consent was
obtained from the patient. Laboratory investigations revealed a
total triiodothyronine value of 1.01 nmol/l (normal range,
0.92–2.79 nmol/l), total thyroxine value of 60.7 nmol/l (normal
range, 58.1–140.6 nmol/l), FT3 value of 3.8 pmol/l (normal range,
3.5–6.5 pmol/l), FT4 value of 12.74 pmol/l (normal range, 11.5–22.7
pmol/l) and a TSH value of 10.01 mU/l (normal range, 0.55–4.78),
indicating subclinical hypothyroidism. The anti-TPOAb and anti-TgAb
assessments were negative. Thyroid ultrasound revealed a normal
sized thyroid gland (right lobe, 3.8×2.0×1.1 cm; left lobe,
3.9×1.9×1.0 cm) with a cystic nodule (2×2.5 cm in diameter) in the
left lobe. A 99mTc scintiscan revealed a cold nodule.
The the patient experienced internal bleeding in the cystic nodule
after feeling a sudden pain in the neck, and was subjected to
subtotal thyroidectomy one week after his admission.
Histologically, the nodule was benign. Immediately after the
thyroidectomy, a tissue sample was obtained from the normal tissue
and was shock-frozen in liquid nitrogen.
Gene sequence analysis
DNA samples were extracted from the patients'
thyroid tissue using a DNA extraction kit (Omega Bio-Tek, Inc.,
Norcross, GA, USA). TSHR gene-coding exons were sequenced by direct
DNA sequencing (Sangon Biotech Co., Ltd., Shanghai, China).
Construction of expression vectors and
site-directed mutagenesis
RNA samples were extracted from the patients'
thyroid tissue using TRIzol (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and the full-length TSHR complementary DNA
(cDNA) was synthesized using the RNA as the template. The primers
used for reverse transcription were as follows: Sense,
5′-CTAAGAGGTACCGAGTCCCGTGGAAAATGA-3′; and antisense,
5′-CCTCTAGACGCCCAACTTACAAAACCGTTTGC-3′ (Sangon Biotech Co. Ltd.).
The reverse transcription reaction mixture included a Takara
Biotechnology Co., Ltd transcription kit (Dalian, China) and the
total volume was 50 µl. The reaction process was performed at 25°C
for 10 min, 37°C for 60 min, then 95°C for 5 min. The mutant TSHR
cDNA fragment was subcloned into a pcDNA3.1 plasmid (Invitrogen;
Thermo Fisher Scientific, Inc.). The mutant TSHR-pcDNA3.1 was used
as a template for mutagenesis to wild-type TSHR plasmid
TSHR-pcDNA3.1 using the Site-Directed Mutagenesis kit (Beijing SBS
Genetech Co., Ltd., Beijing, China) according to the manufacturer's
protocol. All final constructs were verified by direct DNA
sequencing.
Cell culture and in vitro functional
analysis
Mutant and wild-type plasmids were stably
transfected into Chinese hamster ovary (CHO) cells (Chinese Academy
of Sciences, Beijing, China) in Ham's F12 medium with 5% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and
appropriate antibiotics using Lipofectamine reagent enhanced with
Plus Reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. Surviving cells were selected
with 600 mg/l geneticin (G418; Sigma-Aldrich, St. Louis, MO, USA).
A bulk of G418-resistant colonies was used for the subsequent
assessments.
Immunoblot analysis
Crude cell extracts were prepared from the
transfected cells using a protein extraction kit and concentration
was assessed using a bicinchoninic acid assay kit (Beyotime
Institute of Biotechnology, Haimen, China), both in accordance with
the manufacturer's instructions. Following this, 30 µg protein per
sample was separated by 10% SDS-PAGE. Proteins were transferred to
a polyvinylidene fluoride membrane and blocked with 5% non-fat milk
in TBST buffer for 2 h at room temperature with continuous
agitation. Membranes were then incubated with a monoclonal
anti-TSHR antibody (cat. no. MA3-217; Thermo Fisher Scientific,
Inc.) diluted 1:500 and anti-beta actin antibody (cat. no. A01010;
Abbkine, Wuxi, China) diluted 1:5,000 overnight at 4°C. This was
followed by washing and incubation with horseradish
peroxidase-labeled rat anti-mouse immunoglobulin G secondary
antibody (cat. no. IH-0011; Dingguo Changsheng Biotechnology Co.
Ltd, Beijing, China) at a dilution of 1:10,000 in TBST buffer for 2
h at 37°C. The signal was analyzed using enhanced chemiluminescence
detection (Merck Millipore, Darmstadt, Germany). ImageJ software v.
4.18 (imagej.nih.gov/ij/) was used to quantify
the protein
Analysis of competitive TSH
binding
Bovine TSH (bTSH; Sigma-Aldrich) radio-iodination
was performed according to the chloramine-T method as previously
described (4–11). The transfected cells were re-seeded
onto 12-well plates (400,000 cells/well). After 48 h, the medium
was removed and the cells were incubated with 125I-TSH
(1×105 counts/ml; Shanghai Radioimmunity Institute,
Shanghai, China) alone and in the presence of various
concentrations of unlabeled bTSH (0, 0.1, 1, 10, 25, 50, 100, 150,
200, 500 and 1,000 mIU/ml) in the binding buffer (final volume, 0.6
ml) for 1.5 h at 37°C. Subsequently, the buffer was removed, the
cells were lysed with 0.6 ml of 1 M NaOH and the radioactivity was
quantified by a gamma-counter (Multi-Crystal Gamma Counter LB 2111,
Berthold Technologies, Bad Wildbad, Germany) to determine TSH
binding (B).
Analysis of TSH-stimulated cyclic
adenosine monophosphate (cAMP) production
The cells were reseeded onto 96-well plates (25,000
cells/well). After 48 h, the medium was removed and the cells were
incubated with various concentrations of bovine TSH
(0,0.001,0.01,0.1,1,10 and 100 mIU/ml) in the binding buffer (final
volume, 0.1 ml) for 1 h at 37°C. Subsequent to the incubation,
medium was removed and replaced with 0.1 M HCl. The cell extracts
were dried in a vacuum concentrator and cAMP levels were determined
using a cAMP Enzymeimmunoassay kit (Westang Biotechnology Co., Ltd.
Shanghai, China) according to the manufacturer's protocol and
expressed as pmol/ml. Each experiment was performed a minimum of
three times.
Statistical analysis
Data are expressed as the mean ± standard deviation,
from three repetitions. Statistical analysis between the mutation
and wild-type groups was performed using Student's t-test (SPSS
13.0; SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
represent a statistically significant difference.
Results
Identification of V87L mutation in
TSHR gene
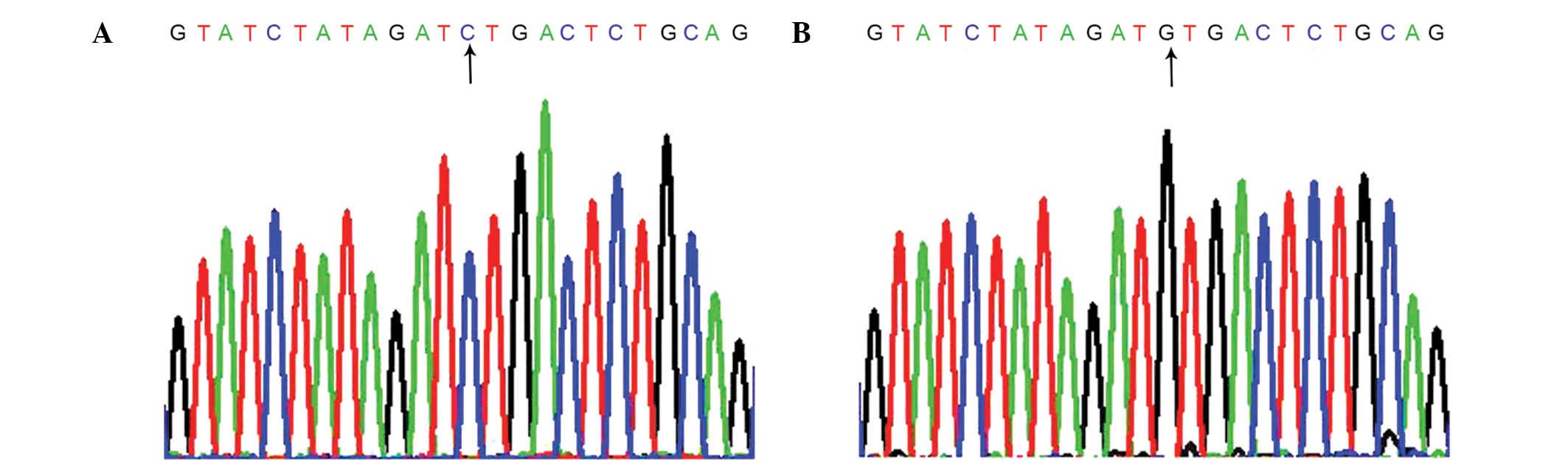
Sequence analysis of the DNA from the patients'
thyroid tissue revealed a guanine-to-cytosine transversion at
position 259 (Fig. 1). The
aforementioned mutation results in an amino acid substitution of
valine by leucine at codon 87 in the extracellular domain.
Functional studies of the novel mutant
receptor
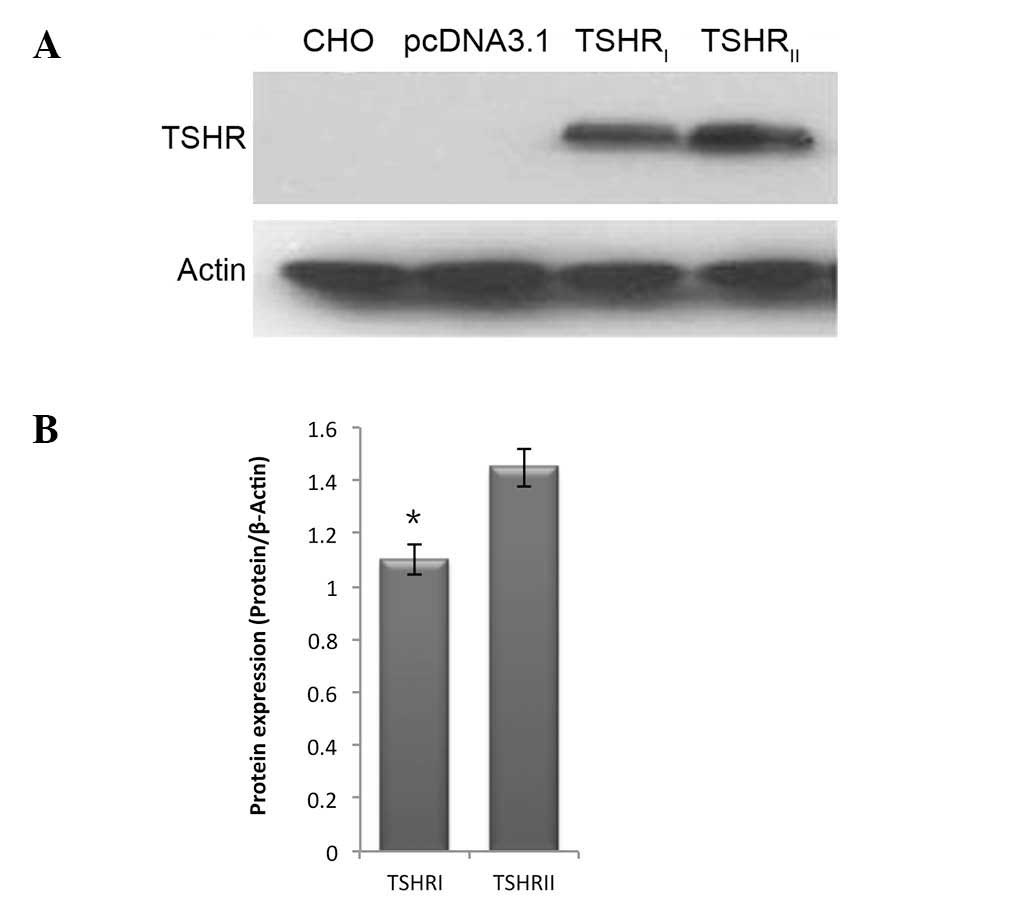
Immunoblot analysis revealed that following
transfection of the ectopic expression plasmids, V87L and the
wild-type TSHR were expressed in CHO cells. However, the V87L
mutant displayed markedly lower expression levels compared with the
wild-type TSHR (Fig. 2).
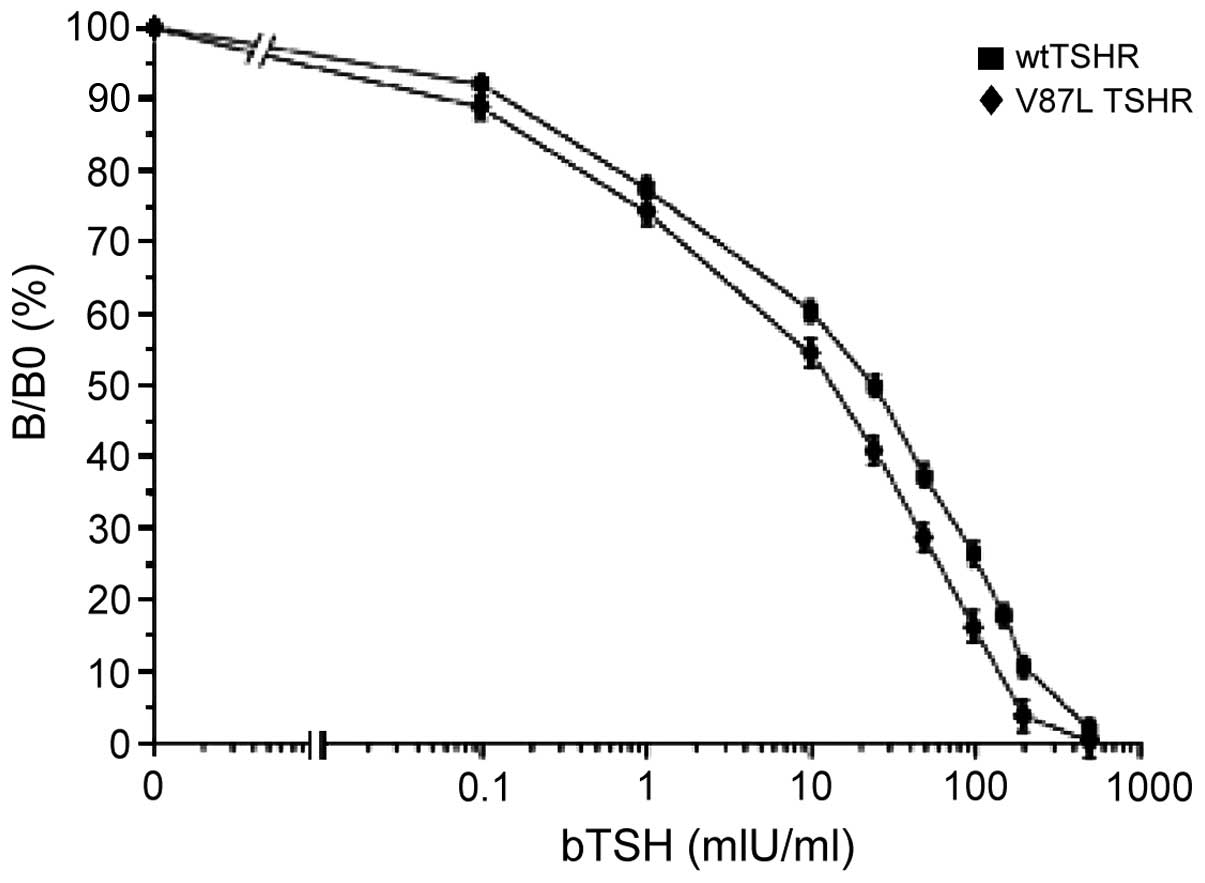
Competitive TSH binding studies
indicate reduced binding activity of the mutant receptor
V87L-TSHR was shown to have a markedly (but not
significantly) reduced TSH binding capacity (B/B0 values) compared
with that of the wild-type TSH receptor (Fig. 3).
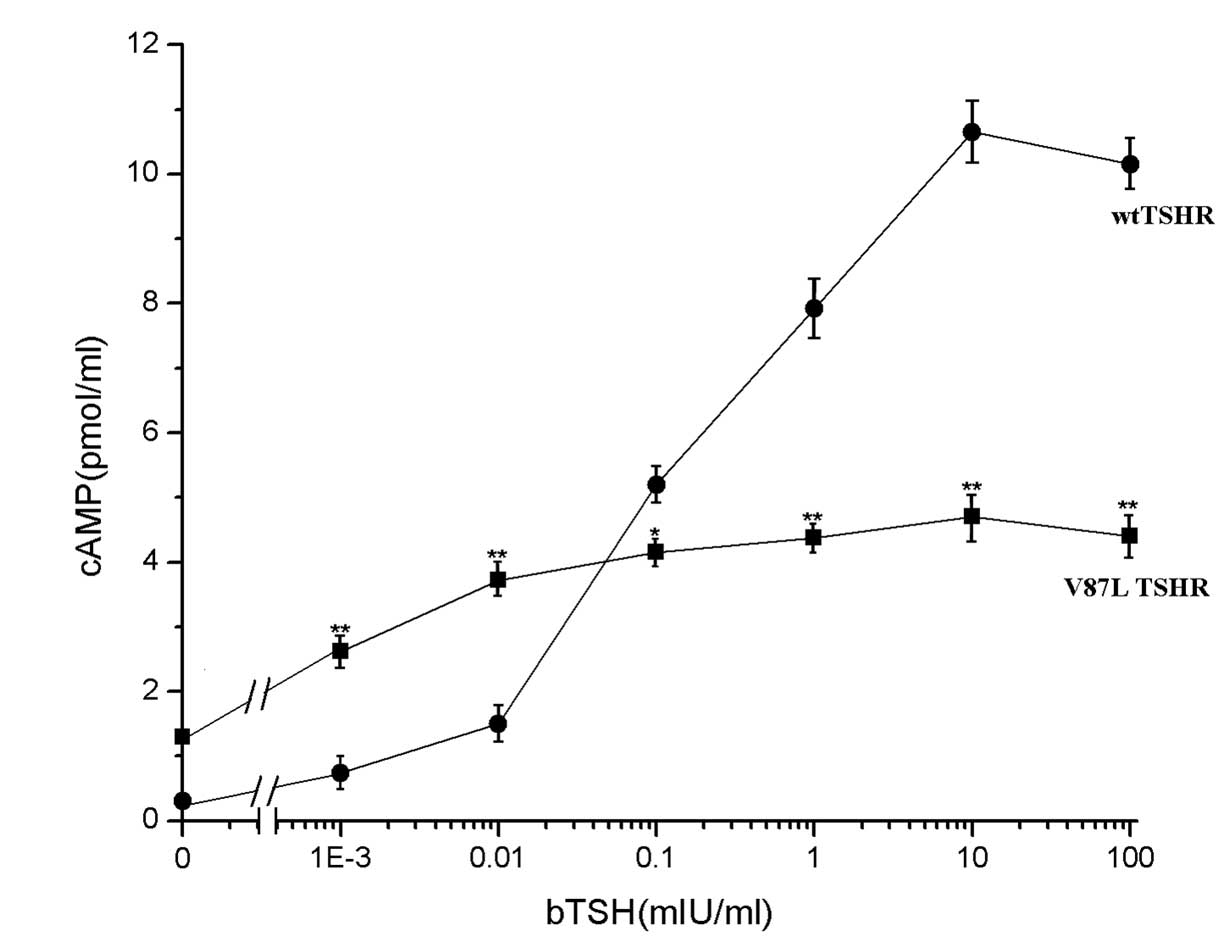
Furthermore, the ability of the CHO cells
transfected with mutant TSHR to produce cAMP in response to bTSH
was significantly decreased. The maximum cAMP concentration did not
exceed half of that in the cells transfected with the wild-type
receptor, and the EC50 values for V87L-TSHR were lower
compared with those for the wild-type receptor (Fig. 4). cAMP production in the
V87L-TSHR-transfected cells showed a gradual increment with the
increase of bTSH concentration, while the cAMP production in the wt
group rose rapidly above a bTSH concentration of 0.01 mIU/ml,
peaking at 10 mIU/ml, indicating the reduced function of the mutant
receptor.
Discussion
The TSHR, located along the basolateral membranes of
the thyroid follicular cells, belongs to the receptor superfamily
of guanine nucleotide-binding protein-coupled receptors, possessing
similar features to other members of the same family, including
follicle-stimulating hormone and luteinizing hormone receptors,
such as the typical structure of seven transmembrane domains. Upon
ligand binding, receptor activation occurs, leading to increased
intracellular cAMP and ultimately thyroid-cell proliferation and
differentiation (15).
The TSHR gene has been defined as highly mutable
(16). Mutations in the TSHR gene
result in either gain or loss of the receptor function.
Gain-of-function mutations are the cause of the following three
syndromes: Familial nonautoimmune hyperthyroidism (FNAH), sporadic
congenital nonautoimmune hyperthyroidism (SCNAH) and autonomous
adenomas (AA) (17). FNAH is also
termed hereditary toxic thyroid hyperplasia or autosomal dominant
autoimmune hyperthyroidism. It is hereditary through a dominant
activating mutation of the TSHR affecting all thyroid cells
(18). SCNAH is a result of germline
neomutations affecting all thyroid cells (19). A large proportion of AAs are also
caused by activating mutations of the TSHR; however, AA affects
only one cell at the clonal origin of a benign tumor (20,21) and
the majority of AA mutations are somatic (22). LOF mutations are predominantly
recessively inherited and lead to the phenotype of TSH
unresponsiveness (2), encompassing a
wide spectrum of transient or permanent clinical and biochemical
manifestations, ranging from complete resistance in nonautoimmune
hypothyroidism with in situ normal-to-hypoplastic thyroid
gland (23–28) or to partial resistance in
nonautoimmune compensated hypothyroidism (2,7–10,29),
defined as slightly elevated serum TSH associated with normal free
thyroid hormone levels. Compensated hypothyroidism is also referred
to as subclinical hypothyroidism (15). Somatic LOF mutations are rare
(4).
The degree of TSH resistance depends on the severity
of the impairment of the receptor function caused by the mutation
and on the number of mutated alleles (30). In cases of both alleles carrying
mutant receptors with complete lack of function, the typical result
is severe congenital hypothyroidism with a hypoplastic thyroid
gland (uncompensated TSH resistance). Less severe LOF mutations,
predominantly recessively inherited, can manifest as
mild/borderline forms of hypothyroidism, in which an appropriate
increase in TSH serum levels can compensates for the reduced
sensitivity of the thyroid (partially or fully compensated TSH
resistance) (14).
The results of the present study were consistent
with the hypothesis that an inactivating mutation of TSHR partially
accounts for nonautoimmune subclinical hypothyroidism. The patient
of the present study had thyroid hormone levels in the lower limit
of normal range and increased serum TSH levels, while she tested
negative for circulating TgAb and TPOAb. The sequence analysis of
the DNA extracted from the patients' thyroid tissue revealed a
guanine-to-cytosine transversion at position 259. This mutation
resulted in an amino acid substitution of valine by leucine at
codon 87 in the extracellular domain. The expression of mutant TSHR
was a little lower than that in the wild-type group; this may be
due to reduced transcription/translation, which requires
confirmation in future studies. Reduced receptor expression may
lead to decreased receptor density at the cell surface, which
causes reduced competitive TSH binding activity. A previous
competitive TSH binding study revealed reduced binding activity of
the mutant receptor. Decreased receptor expression may be a reason
for the reduced TSHR activity, but following binding with TSHR, TSH
functions through cAMP. The cAMP levels in cells transfected with
mutant TSHR vector were decreased compared with those in cells
transfected with wild-type TSHR, indicating that this mutation is
an inactivating mutation. The maximum cAMP concentration in the
mutant-transfected group did not exceed half of that in the
wild-type receptor group, indicating a LOF for the mutated receptor
by >50%.
The results of the sequencing revealed the mutation
to be a homozygous mutant. However, thyroid function was not
completely lost in the patient, although more than half of her
thyroid function was lost. The patient's T4 levels may be
maintained by an appropriate increase in serum TSH levels,
indicating that the mutation of the TSHR did not severely impair
the receptor function. Considering the TSHR gene in its entirety,
this mutational site is a less harmful one. The patient's parents
are deceased and she has no siblings. Her only child, a 34-year-old
male, has a normal thyroid function (TSH, 2.5 mU/l), indicating
that the mutation may be recessively inherited and not X-linked. It
is therefore likely that the mutation is autosomal recessive
inherited.
In conclusion, the present study revealed a novel
TSHR mutation (V87L) in a Chinese woman with subclinical
hypothyroidism, which, to the best of our knowledge, has not been
previously reported. Decreased cAMP accumulation in cells
transfected with V87L-mutated TSHR compared with those transfected
with wild-type receptor suggested that the mutation is associated
with LOF. The findings of the present study may provide valuable
insight into the etiology of SH.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81300642).
References
|
1
|
Vassart G and Dumont JE: The thyrotropin
receptor and the regulation of thyrocyte function and growth.
Endocr Rev. 13:596–611. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Van Sande J, Parma J, Tonacchera M,
Swillens S, Dumont J and Vassart G: Somatic and germline mutations
of the TSH receptor gene in thyroid diseases. J Clin Endocrinol
Metab. 80:2577–2585. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cooper DS: Clinical practice. Subclinical
hypothyroidism. N Engl J Med. 345:260–265. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tonacchera M, Perri A, De Marco G, Agretti
P, Banco ME, Di Cosmo C, Grasso L, Vitti P, Chiovato L and Pinchera
A: Low prevalence of thyrotropin receptor mutations in a large
series of subjects with sporadic and familial nonautoimmune
subclinical hypothyroidism. J Clin Endocrinol Metab. 89:5787–5793.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marcocci C, Vitti P, Cetani F, Catalano F,
Concetti R and Pinchera A: Thyroid ultrasonography helps to
identify patients with diffuse lymphocytic thyroiditis who are
prone to develop hypothyroidism. J Clin Endocrinol Metab.
72:209–213. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Refetoff S: Resistance to thyrotropin.
Endocrinol Invest. 26:770–779. 2003. View Article : Google Scholar
|
|
7
|
Sunthornthepvarakui T, Gottschalk ME,
Hayashi Y and Refetoff S: Brief report: Resistance to thyrotropin
caused by mutations in the thyrotropin-receptor gene. N Engl J Med.
332:155–160. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Roux N, Misrahi M, Brauner R, Houang M,
Carel JC, Granier M, Le Bouc Y, Ghinea N, Boumedienne A, Toublanc
JE and Milgrom E: Four families with loss of function mutations of
the thyrotropin receptor. J Clin Endocrinol Metab. 81:4229–4235.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Clifton-Bligh RJ, Gregory JW, Ludgate M,
John R, Persani L, Asteria C, Beck-Peccoz P and Chatterjee VK: Two
novel mutations in the thyrotropin (TSH) receptor gene in a child
with resistance to TSH. J Clin Endocrinol Metab. 82:1094–1100.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Russo D, Betterle C, Arturi F, Chiefari E,
Girelli ME and Filetti S: A novel mutation in the thyrotropin (TSH)
receptor gene causing loss of TSH binding but constitutive receptor
activation in a family with resistance to TSH. J Clin Endocrinol
Metab. 85:4238–4242. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tonacchera M, Agretti P, De Marco G, Perri
A, Pinchera A, Vitti P and Chiovato L: Thyroid resistance to TSH
complicated by autoimmune thyroiditis. J Clin Endocrinol Metab.
86:4543–4546. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nagashima T, Murakami M, Onigata K,
Morimura T, Nagashima K, Mori M and Morikawa A: Novel inactivating
missense mutations in the thyrotropin receptor gene in Japanese
children with resistance to thyrotropin. Thyroid. 11:551–559. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alberti L, Proverbio MC, Costagliola S,
Romoli R, Boldrighini B, Vigone MC, Weber G, Chiumello G,
Beck-Peccoz P and Persani L: Germline mutations of TSH receptor
gene as cause of nonautoimmune subclinical hypothyroidism. J Clin
Endocrinol Metab. 87:2549–2555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cassio A, Nicoletti A, Rizzello A,
Zazzetta E, Bal M and Baldazzi L: Current loss-of-function
mutations in the thyrotropin receptor gene: when to investigate,
clinical effects, and treatment. J Clin Res Pediatr Endocrinol.
5:(Suppl 1). 29–39. 2013.PubMed/NCBI
|
|
15
|
Camilot M, Teofoli F, Gandini A,
Franceschi R, Rapa A, Corrias A, Bona G, Radetti G and Tatò L:
Thyrotropin receptor gene mutations and TSH resistance: Variable
expressivity in the heterozygotes. Clin Endocrinol (Oxf).
63:146–151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Farid NR, Kascur V and Balazs C: The human
thyrotropin receptor is highly mutable: A review of
gain-of-function mutations. Eur J Endocrinol. 143:25–30. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hébrant A, van Staveren WC, Maenhaut C,
Dumont JE and Leclere J: Genetic hyperthyroidism: Hyperthyroidism
due to activating TSHR mutations. Eur J Endocrinol. 164:1–9. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thomas JS, Leclere J, Hartemann P,
Duheille J, Orgiazzi J, Petersen M, Janot C and Guedenet JC:
Familial hyperthyroidism without evidence of autoimmunity. Acta
Endocrinol (Copenh). 100:512–518. 1982.PubMed/NCBI
|
|
19
|
Kopp P, van Sande J, Parma J, Duprez L,
Gerber H, Joss E, Jameson JL, Dumont JE and Vassart G: Brief
report: Congenital hyperthyroidism caused by a mutation in the
thyrotropin-receptor gene. N Engl J Med. 332:150–154. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Corvilain B, Van Sande J, Dumont JE and
Vassart G: Somatic and germline mutations of the TSH receptor and
thyroid diseases. Clin Endocrinol (Oxf). 55:143–158. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krohn K, Führer D, Holzapfel HP and
Paschke R: Clonal origin of toxic thyroid nodules with
constitutively activating thyrotropin receptor mutations. J Clin
Endocrinol Metab. 83:130–134. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eszlinger M, Niedziela M, Typlt E,
Jaeschke H, Huth S, Schaarschmidt J, Aigner T, Trejster E, Krohn K,
Bösenberg E and Paschke R: Somatic mutations in 33 benign and
malignant hot thyroid nodules in children and adolescents. Mol Cell
Endocrinol. 93:39–45. 2014. View Article : Google Scholar
|
|
23
|
Abramowicz MJ, Duprez L, Parma J, Vassart
G and Heinrichs C: Familial congenital hypothyroidism due to
inactivating mutation of the thyrotropin receptor causing profound
hypoplasia of the thyroid gland. Clin Invest. 99:3018–3024. 1997.
View Article : Google Scholar
|
|
24
|
Biebermann H, Schöneberg T, Krude H,
Schultz G, Gudermann T and Grüters A: Mutations of the human
thyrotropin receptor gene causing thyroid hypoplasia and persistent
congenital hypothyroidism. J Clin Endocrinol Metab. 82:3471–3480.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gagné N, Parma J, Deal C, Vassart G and
Van Vliet G: Apparent congenital athyreosis contrasting with normal
plasma thyroglobulin levels and associated with inactivating
mutations in the thyrotropin receptor gene: Are athyreosis and
ectopic thyroid distinct entities? J Clin Endocrinol Metab.
83:1771–1775. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tiosano D, Pannain S, Vassart G, et al:
The hypothyroidism in an inbred kindred with congenital thyroid
hormone and glucocorticoid deficiency is due to a mutation
producing a truncated thyrotropin receptor. Thyroid. 9:887–894.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tonacchera M, Agretti P, Pinchera A, et
al: Congenital hypothyroidism with impaired thyroid response to
thyrotropin (TSH) and absent circulating thyroglobulin: Evidence
for a new inactivating mutation of the TSH receptor gene. J Clin
Endocrinol Metab. 85:1001–1008. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jordan N, Williams N, Gregory JW, Evans C,
Owen M and Ludgate M: The W546X mutation of the thyrotropin
receptor gene: Potential major contributor to thyroid dysfunction
in a Caucasian population. J Clin Endocrinol Metab. 88:1002–1005.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Calaciura F, Motta RM, Miscio G, Fichera
G, Leonardi D, Carta A, Trischitta V, Tassi V, Sava L and Vigneri
R: Subclinical hypothyroidism in early childhood: A frequent
outcome of transient neonatal hyperthyrotropinemia. J Clin
Endocrinol Metab. 87:3209–3214. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Persani L, Calebiro D, Cordella D, Weber
G, Gelmini G, Libri D, de Filippis T and Bonomi M: Genetics and
phenomics of hypothyroidism due to TSH resistance. Mol Cell
Endocrinol. 322:72–82. 2010. View Article : Google Scholar : PubMed/NCBI
|