Introduction
Langerhans' cell histiocytosis (LCH) is a rare
disease with variable clinical appearance. The etiopathogenesis of
LCH is still unclear and remains to be investigated. Despite the
fact that it had been widely discussed that viral or genetic
predisposing factors make serious contributions to this disease, no
conclusive proof has ever been provided (1). In 1998, Weintrarb et al
(2) demonstrated that p53 was
detected in all LCH biopsy specimens, particularly in Langerhans
cells; according to their report, the inactivation of p53 led to an
uncontrolled proliferation of LCH cells. The annual incidence of
LCH is ~5.4 cases per one million people, and a male predominance
could be observed. LCH is primarily a pediatric disease, which is
rarely reported in adults. Moreover, the peak age of the patients
range between 1 and 4 years old, and patients with focal lesions
are generally older (0.1–15.1 years) than those with multisystemic
disease (0.09–14.8 years) (3).
Histiocytosis of the temporal bone occurs in 15–60% of the sample
cases, and it is higher when radiological findings are included in
asymptomatic patients; its bilateral occurrence is described in up
to 30% of the cases (4) and is more
frequent in the multisystemic diseases (5,6). The
characteristics of LCH are local invasive growth that easily
relapses following treatment, and possible disseminated malignant
tumors (7). Moreover, the disease
has complicated clinical manifestations of which bone damage is the
most common (8). The clinical
features, diagnosis, treatment and prognosis of the LCH remain
obscure, and it is easy to confuse temporal LCH with ear
inflammatory lesions and malignant tumors (8). Thus, the aim of the present study is to
illustrate the clinical presentations, diagnosis, management and
prognosis of this disease.
Case report
A 20-month-old Chinese boy presented with a 1-month
history of worsening left facial paralysis and imbalance and
progressive hearing loss, which co-presented with purulence of the
left external auditory meatus and a slowly enlarging post auricular
mass, swelling of the ear and the temporal bone. Free-field
pure-tone audiometry showed moderate conductive deafness and there
was no pathological nystagmus. The blood routine examination
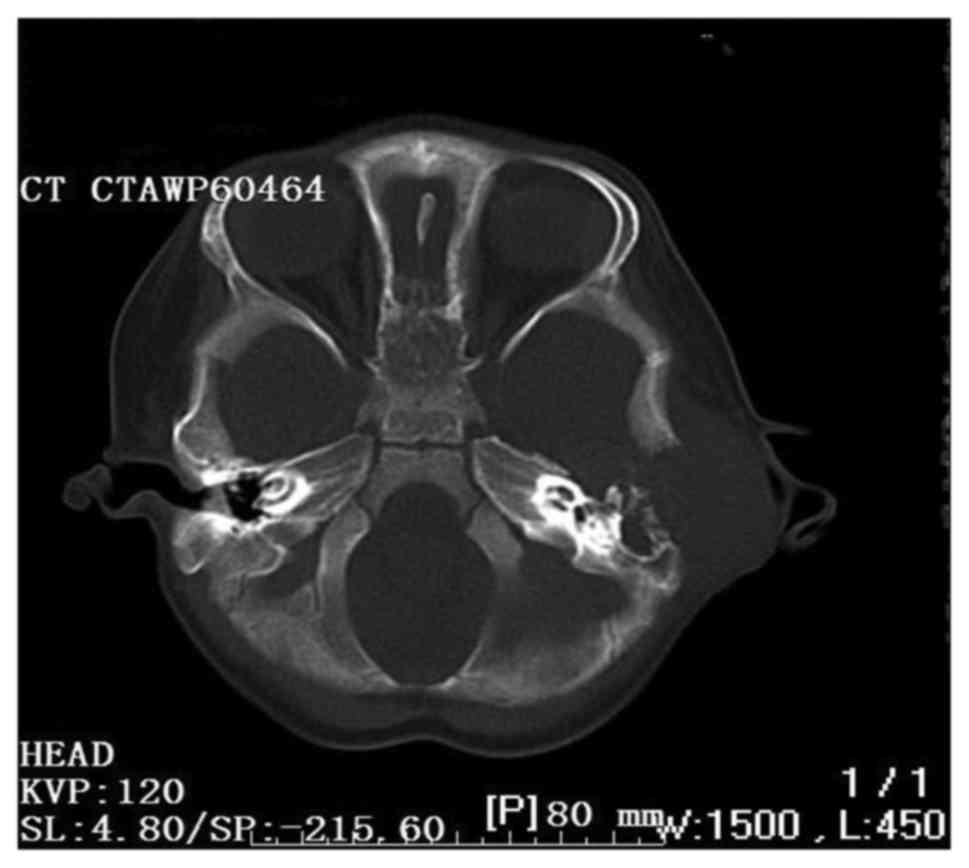
revealed leukocytosis (27.2/ml). The computed tomography scan
revealed a destructive bone lesion involving the left posterior
external canal, middle and posterior cranial fossa, and the lateral
semicircular canal (Fig. 1).
The cutaneous infiltrates were located in the
papules of the mid-line of the trunk and the flexor areas of the
limbs. Moreover, radiological images demonstrated a micronodular
infiltrate of the lungs and evidence of nodes that were revealed in
the liver. The levels of bilirubin and aminotransferases in the
serum were also increasing. The patient was initially diagnosed
with malignant granuloma of the ear or inflammatory lesions of the
ear. Moreover, surgical biopsies were obtained via a mastoidectomy
approach. During surgery, vast destruction of the temporal bone
involving the left posterior external canal, middle and posterior
cranial fossa, and the lateral semicircular canal were confirmed,
and fibrous inflammatory tissue interrupted by necrotic and
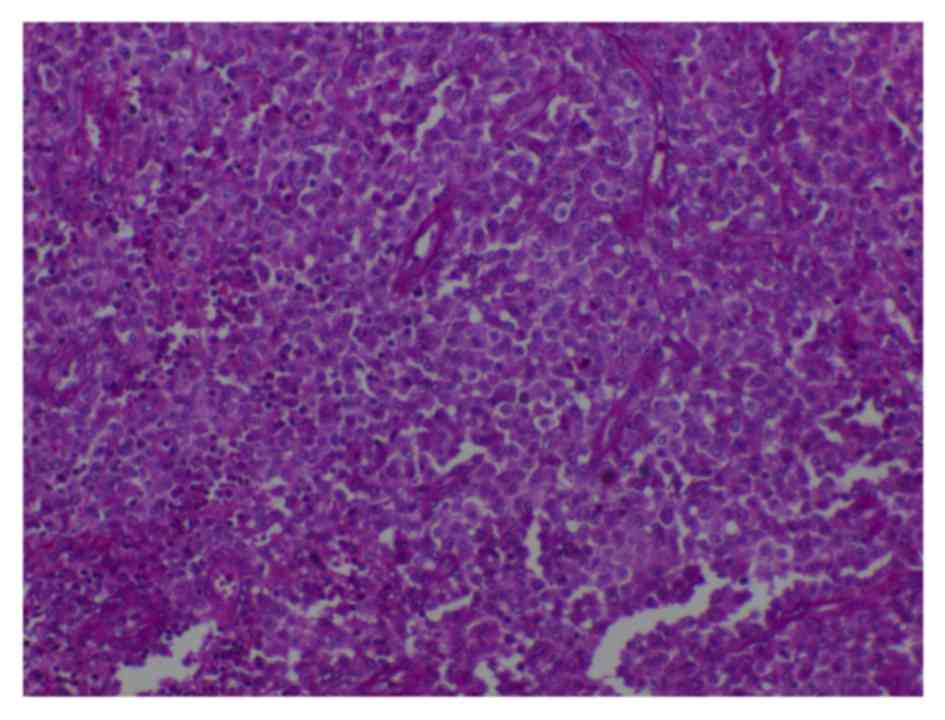
purulent material was found. Intraoperative histology revealed bony
and fibrotic tissue with interposed infiltrate containing
lymphocytes, eosinophilic granulocytes, and typical Langerhans
histiocytes. A final diagnosis of LCH was made by a surgical
pathological examination that revealed numerous ovoid
Langerhans' histocytic cells with grooved and lobulated
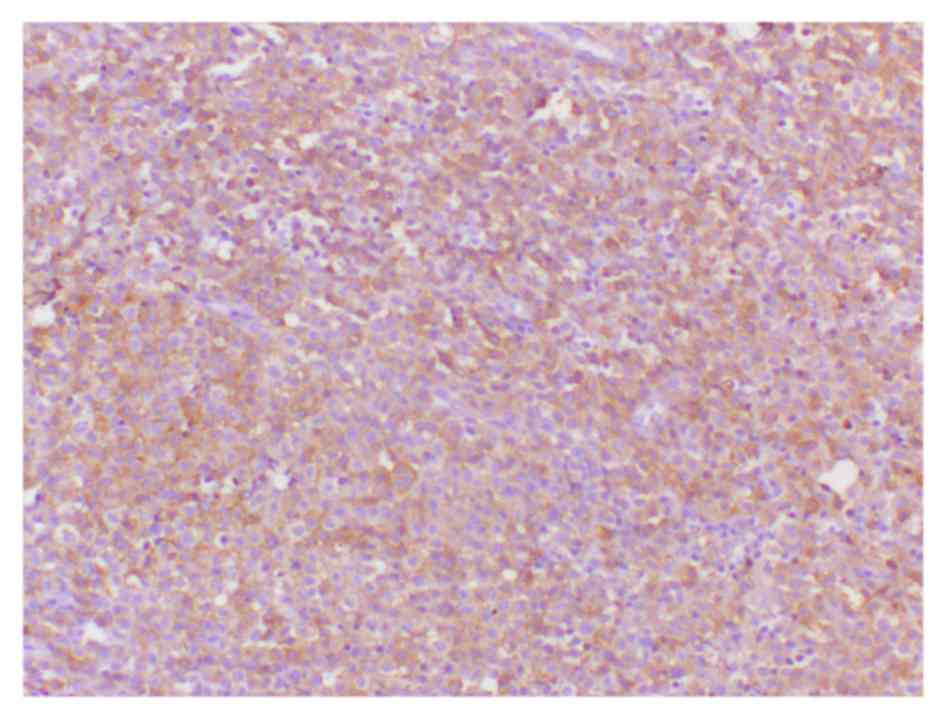
nuclei and fine chromatin, which were positive for CD1/S-100 by
immunohistochemical staining (Figs.
2 and 3). Following surgery,
further chemotherapy was planned, however the parents of the boy
refused any additional therapy and removed him from the hospital,
and the boy succumbed to the disease 6 months later.
Discussion
LCH is a rare disorder that is associated with a
wide spectrum of presenting symptoms and variable clinical behavior
and prognosis. In addition to the temporal bone, the head and neck
area involving the orbital bone, the scalp, the mandible, the
maxillary bone and the facial skin may be involved when LCH is
evident. Clinical manifestations varied depending on the site of
involvement; thus, there is no characteristic pathognomonic
presentation that specifically identifies LCH (9). The classical disease is divided into
three overlapping states called eosin ophitic granuloma,
Hand-Schüller-Christian disease, and Letterer-Siwe disease
(10).
LCH is capable of affecting reticuloendothelial
cells anywhere in the body, and as such, multiple organs may be
involved.
The regions that are most frequently involved
include the bones, skin, liver, lung, spleen, hypophysis, lymph
nodes and bone marrow (11).
Moreover, cutaneous lesions are observed in almost 50% of patients,
and can be the only sign of the disease or evidence of a
multisystem involvement. Rash is one of the most common effects,
and the preferred location is the mid-line of the trunk and the
flexor areas of the limbs. Furthermore, the scalp has erythematous
lesions that may evolve into petechiae that ulcerate and form
crusts (12).
Pulmonary involvement is observed in 20–40% of
patients and manifests clinically as a cough, dyspnea and tachypnea
amongst other presentations (13).
In addition, radiological images commonly reveal a micronodular
presence of cystic infiltrates. The main complication of
gastrointestinal lesions is bleeding (13). In addition, the levels of
aminotransferases and bilirubin increase when hepatic cells are
involved (13). Moreover, the
hematological alterations may indicate a lesion in the bone marrow
or in the spleen. Enlarged cervical lymph nodes are affected either
in a single isolated organ presentation of the disease or as part
of a multisystem involved disease (14).
LCH can also be easily misdiagnosed because of the
common involvement of the external and middle ear (15). A previous study has reported a
misdiagnosis rate of ~72.7% (15/22) for LCH, primarily due to the
clinical presentation resembling that caused by other more common
conditions (15). Moreover,
diagnosis of LCH should be based on structured and synthetic
analyses of the clinical manifestations, the clinical features of
imaging and their histopathology. Pathologically, LCH is
characterized by multinucleated Langerhans' cells, histiocytes and
eosinophils, despite the hallmark cell of LCH being Langerhans'
cell histiocyte (16). The gold
standard for the diagnosis of the disease requires the presence of
Birbeck granules on an electron microscopic examination; however,
Birbeck granules are not exclusively observed in this disease,
since they have already been identified in other inflammatory
conditions of the lymph nodes (16).
However, in certain cases, immunohistochemistry can be fundamental
in establishing a diagnosis. For example, a diagnosis can be
established with the use of the cell surface or cytoplasmic
expression of CD1a, S100 and/or CD45 immunostaining on
histopathological specimens (17).
According to the diagnostic criteria of the
international organization cell association for LCH in 1987,
diagnosis of LCH was divided into three degrees of staging.
Firstly, the preliminary diagnosis relies on the results of the
clinical laboratory, including bone and lung X-rays and common
pathological changes (e.g., abnormal Langerhans cells are shown to
be present in pathological sections). Secondly, the diagnosis can
be made if analysis by immunohistochemistry demonstrates a positive
result for S-100 protein based on the preliminary diagnosis.
Third-degree diagnosis is made from a detailed examination of the
preliminary diagnosis, along with ultrastructural examination that
shows Birbeck particles or a positive result for CD1a expression by
immunohistochemistry (18).
An ideal therapy for LCH has not yet been
established. However, the objectives of the treatment include
improvement of clinical symptoms and prevention of complications
(19). There are several treatment
modalities for temporal bone LCH, including surgery, radiotherapy,
steroidal injections and chemotherapy. In addition, they can be
used alone or in combination depending on the severity and extent
of the disease (20). It is
noteworthy that ~10–20% of patients have revealed a spontaneous
regression of the disease. Moreover, it is accepted that, if
possible, a surgical resection of the lesions is preferred as a
treatment. Nevertheless, a number of authors prefer the application
of chemotherapy. Different protocols, in addition to the use of
systemic corticosteroids, cytostatic therapies like
2-chlorodesoxiademosin (2CdA-cladribine), cyclosporine, thalidomide
and myeloablative therapies were prescribed (19,21–23).
Single system disease can be treated with local injectable
intratumoral steroids (11,18). Following surgery, numerous
investigators recommend low-dose radiotherapy (10–20 Gy) in cases
in which the tissue involved cannot be completely excised (21,24).
Although LCH is very critical, the prognosis is
generally good if vital organs are not involved and correct
diagnosis and a timely rational therapy can be made. The indices of
response, recurrence and complications may vary due to different
schemes. Moreover, the prognosis of temporal bone LCH is excellent
in patients with limited organ involvement (3). Besides the extent of the disease, the
age of the patient at the time of diagnosis is an important factor.
In addition, children <2 years of age have a worse outcome. In
addition, the presence of cervical lymph nodes and scalp
involvement presents as a poorer prognosis and an indicator for
recurrence. Finally, the most relevant prognostic factors are
multisystem involvement and vital organ dysfunction. In general,
the patients with target organ lesions have a worse outcome and a
higher incidence of complications and risk of death (25).
In conclusion, the case of this young male patient
described in the present case report <2 years of age and
suffered from temporal bone LCH. In addition, the patient also
suffered from multiple organ failure including that of the liver,
kidney, lymphatic system, skin and hematopoietic system. Therefore,
patients with lesions in target organs have worse outcomes and a
higher incidence of complications and mortality.
References
|
1
|
Campos MK, Viana MB, de Oliveira BM,
Ribeiro DD and Silva CM: Langerhans cell histiocytosis: A 16-year
experience. J Pediatr (Rio J). 83:79–86. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weintraub M, Bhatia KG, Chandra RS,
Magrath IT and Ladisch S: p53 expression in Langerhans cell
histiocytosis. J Pediatr Hematol Oncol. 20:12–17. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saliba I, Sidani K, El Fata F, Arcand P,
Quintal MC and Abela A: Langerhans' cell histiocytosis of the
temporal bone in children. Int J Pediatr Otorhinolaryngol.
72:775–786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cochrane LA, Prince M and Clarke K:
Langerhans' cell histiocytosis in the paediatric population:
Presentation and treatment of head and neck manifestations. J
Otolaryngol. 32:33–37. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
DiNardo LJ and Wetmore RF: Head and neck
manifestations of histiocytosis-X in children. Laryngoscope.
99:721–724. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Irving RM, Broadbent V and Jones NS:
Langerhans' cell histiocytosis in childhood: Management of head and
neck manifestations. Laryngoscope. 104:64–70. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kleinjung T, Woenckhaus M, Bachthaler M,
Wolff JE and Wolf SR: Langerhans' cell histiocytosis with bilateral
temporal bone involvement. Am J Otolaryngol. 24:265–270. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vezina JP, Audet N and Fradet G:
Cerebrospinal fluid otorrhoea: A rare presentation of Langerhans'
cell histiocytosis of the temporal bone. J Laryngol Otol.
124:545–548. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grant GA, Kim DK, Shaw CM and Berger MS:
Solitary eosinophilic granuloma of the temporal lobe: Case report
and review of the literature. Brain Tumor Pathol. 16:55–59. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Isaacs H Jr: Fetal and neonatal
histiocytoses. Pediatr Blood Cancer. 47:123–129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ha SY, Helms P, Fletcher M, Broadbent V
and Pritchard J: Lung involvement in Langerhans' cell
histiocytosis: Prevalence, clinical features, and outcome.
Pediatrics. 89:466–469. 1992.PubMed/NCBI
|
|
12
|
Nanduri VR, Pritchard J, Chong WK, Phelps
PD, Sirimanna K and Bailey CM: Labyrinthine involvement in
Langerhans' cell histiocytosis. Int J Pediatr Otorhinolaryngol.
46:109–115. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ha SY, Helms P, Fletcher M, Broadbent B
and Pritchard J: Lung involvement in Lanerhans' cell histiocytosis:
Prevalence, clinical features, and outcome. Pediatrics. 89:466–469.
1992.PubMed/NCBI
|
|
14
|
Chen L, Wang WQ, Xu H and Chi FL:
Langerhans cell histiocytosis of the temporal bone: 22 cases
analysis. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi.
45:212–216. 2010.(In Chinese). PubMed/NCBI
|
|
15
|
Brown CW, Jarvis JG, Letts M and Carpenter
B: Treatment and outcome of vertebral Langerhans cell histiocytosis
at the Children's Hospital of Eastern Ontario. Can J Surg.
48:230–236. 2005.PubMed/NCBI
|
|
16
|
Herwig MC, Wojno T, Zhang Q and
Grossniklaus HE: Langerhans cell histiocytosis of the orbit: Five
clinicopathologic cases and review of the literature. Surv
Ophthalmol. 58:330–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kasper EM, Aguirre-Padilla DH, Alter RY
and Anderson M: Histiocytosis X: Characteristics, behavior, and
treatments as illustrated in a case series. Surg Neurol Int.
2:572011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Barros Fernandes Humberlo, Granjeiro RC
and de Negreiros Júnior J: Langerhans Cell Histiocytosis in
Otorhinolaryngology. 13:444–449. 2009.
|
|
19
|
Bayazit Y, Sirikci A, Bayaram M, Kanlikama
M, Demir A and Bakir K: Eosinophilic granuloma of the temporal
bone. Auris Nasus Larynx. 28:99–102. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mosnier I, Rondini-Gilli E, Crosara PT,
Belmatoug N, Cyna-Gorse F, Cazals-Hatem D, Abbey-Toby A,
Bozorg-Grayeli A and Sterkers O: Langerhans' cell histiocytosis of
the labyrinth in adults. Otol Neurotol. 25:27–32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Martini A, Aimoni C, Trevisani M and
Marangoni P: Langerhans' cell histiocytosis: Report of a case with
temporal localization. Int J Pediatr Otorhinolaryngol. 55:51–56.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu JJ, Huang DB, Pang KR, Hsu S and Tyring
SK: Thalidomide: Dermatological indications, mechanisms of action
and side-effects. Br J Dermatol. 153:254–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dornfeld S, Winkler C, Dörr W and Herrmann
T: The radiotherapy of histiocytosis X. A case report of an
eosinophilic granuloma in adulthood and a review of the literature.
Strahlenther Onkol. 174:534–535. 1998.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Heyd R, Strassmann G, Donnerstag F, Martin
T and Zamboglou N: Radiotherapy in Langerhans-cell histiocytosis. 2
case reports and review of the literature. Rontgenpraxis. 53:51–61.
1999.(In German).
|
|
25
|
Howarth DM, Gilchrist GS, Mullan BP,
Wiseman GA, Edmonson JH and Schomberg PJ: Langerhans cell
histiocytosis: Diagnosis, natural history, management, and outcome.
Cancer. 85:2278–2290. 1999. View Article : Google Scholar : PubMed/NCBI
|