Introduction
Hypertrophic cardiomyopathy (HCM) is a common
autosomal dominant cardiac disease characterized by asymmetric left
ventricular (LV) hypertrophy (LVH) with heterogeneous morphologic,
functional and clinical features (1). Since the pathogenic missense mutation
in the β-myosin heavy chain 7 (MYH7 R403Q) was revealed two
decades ago, over 1,400 gene mutations have been identified in ≥11
putative HCM-susceptibility genes (2–4), which
predominantly encode for proteins of the thick filament of the
cardiac sarcomere. Cardiac myosin-binding protein C3
(MyBPC3) and myosin heavy chain 7 (MYH7) are, by far,
the most common HCM-associated genes with an estimated prevalence
of 25–35% for each gene (5). MyBPC3
is a key constituent of the thick filaments localized to doublets
in the C-zone of the A-band of the sarcomere. By binding to myosin,
titin and actin, MyBPC3 contributes to the structural integrity of
the cardiac sarcomere and regulates cardiac muscle contractility
(6).
Although the mechanisms underlying the correlations
between disease-causing genetic mutations and characteristic
pathological and morphologic (or phenotypic) features of HCM are
unclear, preliminary studies suggest that particular gene
abnormalities are associated with specific clinical phenotypes,
such as degree of hypertrophy, risk of sudden death, onset of
disease and disease penetrance in families (7–9).
Mutations in the MyBPC3 gene were found to be responsible
for 15–20% of cases of familial HCM, and were generally associated
with mild and age-related penetrance disease (10). However, the prevalence of
MyBPC3 mutations and the clinical characteristics associated
in the Chinese HCM population remain unclear.
The present study is, to the best of our knowledge,
the first to identify the prevalence of MyBPC3 gene
mutations and the associated characteristic echocardiographic
phenotypes in Chinese patients with HCM. To identify these clinical
characteristics, cutting-edge echocardiographic techniques, such as
2-dimensional echocardiography (2DE), Tissue Doppler imaging (TDI)
and real-time 3-dimensional echocardiography (RT-3DE), were
employed.
Materials and methods
Study population
A total of 48 Chinese adult patients (age, 48±12
years) with familial or sporadic HCM were enrolled in the study
after informed written consent was obtained. All patients were in
sinus rhythm at the time of enrollment and were diagnosed
clinically by echocardiography between December 2008 and February
2010 at the General People's Liberation Army Hospital in Beijing,
China. A total of 37 patients had a family history of HCM while the
other 11 patients did not have confirmed relatives with HCM. The
diagnosis of HCM was based on 2DE showing an unexplained maximum
left ventricular wall thickness (MLVWT) of ≥15 mm (11) in the absence of other cardiac or
systemic disease capable of producing the observed magnitude of
hypertrophy (such as systemic hypertension, aortic stenosis and
amyloidosis). The family members of index patients with positive
MyBPC3 mutations were divided into the following two groups
according to whether the patients and their family members met the
criteria for diagnosis of HCM from the results of routine
echocardiography, as described by McKenna et al (12): Mutation carriers in the LVH group
(G+/LVH+; n=18), and mutation carriers not in the LVH group
(G+/LVH-; n=23). The control group consisted of 30 healthy age- and
gender-matched subjects without cardiological disorders.
Genetic study
Venous blood (3 ml) was collected from each subject
for MyBPC3 mutation screening. Systematic family screenings
of genotyped patients with HCM were performed after identifying
MyBPC3 mutations. DNA was extracted from whole blood and
stored at −70°C. Genomic primer pairs were designed to amplify the
whole coding exons of MyBPC3 (35 exons) using Primer BLAST
(www.ncbi.nlm.nih.gov/tools/primer-blast/) and Primer
Premier 5 software (PREMIER Biosoft, Palo Alto, CA, USA). Nested
polymerase chain reaction (PCR) was used to amplify the extracted
DNA (Table I). Briefly, for the
initial PCR, 0.1 µg DNA and primers were used in a 20-µl reaction:
2.5 µl 10X buffer, 0.5 µl 2.5 mM dNTPs, 0.4 µl of each primer
forward (10 µM) and reverse (10 µM) and 2.5 µl of Taq (TaKaRa) (5
U/µl). The target gene was amplified according to the following
parameters: Denaturation at 95°C for 5 min, 45 cycles of
denaturation at 95°C for 25 sec, annealing at 50°C for 60 sec and
extension at 72°C for 2 min, and a final extension of 5 min at
72°C. The second round of PCR was performed using the first-round
PCR products in a 20-µl reaction: 2.5 µl 10X buffer, 0.5 µl 2.5 mM
dNTPs, 0.5 µl of each primer forward (10 µM) and reverse (10 µM)
and 2.5 µl of Taq (TaKaRa) (5 U/µl). The target gene was amplified
according to the following parameters: denaturation at 95°C for 3
min, 30 cycles of denaturation at 95°C for 30 sec, annealing at
55°C for 45 sec and extension at 72°C for 45 sec, and a final
extension of 2 min at 72°C. Following PCR amplification, PCR
products were analyzed by direct sequencing on an ABI PRISM 3130
DNA Analyzer with BigDye Terminator chemistry (version 3.1; Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
fluorophore used was SYBR green (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The 2−ΔΔcq method was used to
quantify the results (13). All
results were repeated four independent experiments. The relative
level of mRNA was calculated by the comparative CT method with
GAPDH mRNA as the housekeeping gene.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Forward primer
(5′–3′) | Reverse primer
(5′–3′) |
|---|
| Exon1 |
GAGGCAGATAAGCAGAGCCT' |
CCCTCAAGAACTCCCTCCT |
| Exon2 |
GCATAGAAAGTGCTAGCACA |
GGAAGGCTGATCAGGATCTT |
| Exon3 |
GACAGCCATGGCAGACTTT |
TTGAGACCTGCCCTGGACA |
| Exon4 |
CCTTTGCTCACAGGGTCAA |
CATTTGCCCTTGAACCACT |
| Exon5 |
CCATTGGCCTCTTCGTGAT |
CATTTGCCCTTGAACCACT |
| Exon6 |
CCCAGTCTCCTTTAAGGGT |
GAGGCATCCTCCTTAGTGTT |
| Exon7 |
GAATGGGCAAGTCTGTGAAT |
CTCAGTATCCTCACCTGCCT |
| Exon8 |
GGCAGGTGAGGATACTGAGT |
GAAAGGGACACTAGCCAGAT |
| Exon9 |
CAATCTGGCTAGTGTCCCTT |
GACTGTTGACGGGACATAAT |
| Exon11 |
GGTGGCCATACCTCTCATGT |
TAGGATCTCCCACACGTCCT |
| Exon12 |
GCTACAGCTCCTTGGTCCT |
GTGTAGGGAAGGGCTAGCCT |
| Exon13 |
GGTGCTCAGCCTTTCAGAA |
CGAGTCAGAGATACGCATGT |
| Exon15 |
GCAGCTTTCCTGCCACTTC |
GTGAGCATGAGGGTTGGCT |
| Exon16 |
CCTGAGGATGTGGGAACCT |
CAAGTGCTGTGGCCTCTTCT |
| Exon17 |
GCGCAAGTCAAATGGTGAGT |
CAAGCCCTAAAGCCTCATGT |
| Exon18 |
CTCAGACACTTGAGGTTCCT |
CTCTGTCTCCATCTCAGTCT |
| Exon19 |
CCAACAAGCCAGGACAAGGT |
CGGGAAAGTGAGCAGAACCA |
| Exon 20 |
CCAACAAGCCAGGACAAGGT |
CGGGAAAGTGAGCAGAACCA |
| Exon21 |
CTCTCCCGTTTCTCTGAACT |
GGTTCCACACACCCATCTTA |
| Exon22 |
CTGAGTCAGCTCCTCTGCT |
TGATGGCCATCAGCACACT |
| Exon23 |
GAACTAGATGCTGACGTGGA |
GTTTGTCGAGTGGCTGAAT |
| Exon24 |
GCGGTTAGTTGGAGTGGGA |
CATCCACCGGTAGCTCTTCT |
| Exon25 |
GGACTCCTGCACAGTACAGT |
CCTGCAGAGCACCTGCTATT |
| Exon26 |
CTATGTGACCAGTGGGCAGT |
CTCTGGGTGTCCTCAACTTT |
| Exon27 |
GTCAGTGGTGACACAGCCT |
GGGTCTTGTGACTGCACAAA |
| Exon28 |
GTGTTAGCAGGAGCTAAAGG |
CTGGATGGGAACAACACACT |
| Exon29 |
GAGTGATCCAGGTTCAGGGT |
GGAAAATGTGAGCTGTGGGT |
| Exon30 |
CCAACCCACAGCTCACATTT |
GAGGACAGTGAAGGGTAGCT |
| Exon31 |
GCTGATCTGAATCCCTCCAT |
CTGGTTGGAAGAATGAGGGT |
| Exon32 |
TCTCGGTACCAAGTCCTGT |
GGAACCAAGAGTGAGTACCA |
| Exon33 |
CTCTCAGCCTGGATGGCTT |
CCGAGGACAACGGAGCAAA |
| Exon34 |
GCAATAGCTTCCAGAAGGCT |
CCTCCCATTTACTGATGGCT |
| Exon35 |
CATCAGTAAATGGGAGGCTG |
GGCACACCGAAATTGAGAA |
For every sequence variant detected, a cohort of 200
ethnically matched control subjects were screened by using the same
methodology. Conservation of residues was determined by Homologene
(www.ncbi.nlm.nih.gov/homologene) by multiple alignment
of orthologues in various species, including Homo sapiens,
Pan troglodytes, Canis lupus, Bos taurus,
Mus musculus, Rattus norvegicus, Gallus gallus
and Danio rerio. A variant was considered a mutation if it
showed co-segregation with affected members in the family, was not
present in the 200 healthy adult controls, and if the mutated
residue was conserved among species.
Clinical evaluation
Index patients and their relatives were evaluated
for family history, age, gender, body surface area and clinical
symptoms. They also underwent 12-lead electrocardiography, 2DE,
TDI, and 3DE.
2-DE
2DE images were acquired with a commercially
available ultrasound system (Acuson Sequoia 512; Siemens AG,
Munich, Germany) equipped with a 4V1c transducer (1–4 MHz). LV
end-diastolic diameter (LVEDD) and LV end-systolic diameter (LVESD)
were measured using M-mode and 2D images obtained from parasternal
long-axis views. The severity and distribution of LVH was assessed
in the parasternal short-axis plane at the mitral valve and
papillary muscle levels. MLVWT was regarded as the greatest
thickness at any site in the LV wall (14). Patients were classified as having
obstructive HCM when the LV outflow tract gradient was ≥30 mmHg
(15). The mitral early (E-wave) and
late (A-wave) filling velocities ratio (E/A), and the E-wave
velocity deceleration time (DT) were measured by pulsed-wave
Doppler. TDI was performed by placing the sample volume at the side
of the basal septal and lateral wall in the apical 4-chamber view.
The mitral annular systolic (Sa), and early (Ea) and late (Aa)
diastolic velocities were recorded at late expiration in the
pulsed-wave Doppler mode. Diastolic function was subsequently
graded 0 to 3, as previously described (16): Grade 0 (normal relaxation): DT
>140 msec, E/A ratio >1 and septal E/Ea ratio <8; Grade 1
(impaired relaxation): DT >140 msec, E/A ratio <0.75 and E/Ea
ratio <8; Grade 2 (pseudo-normal filling): DT >140 msec, 1
<E/A ratio <1.5 and E/Ea ratio >15, and systolic/diastolic
pulmonary vein ratio <1; Grade 3 (restrictive filling): E/A
ratio ≥1.5, DT <140 msec, E/Ea ratio >15, and
systolic/diastolic pulmonary vein ratio <1.
3-DE
3DE images were obtained with a commercially
available ultrasound system (Acuson SC2000; Siemens AG) equipped
with a 4Z1c transducer (2–4 MHz). This system has 15–17 cm
detectable depth, 90×90° scanning angle, and >20 frames/sec
volume rate. Electrocardiogram (ECG) images were obtained in the
left lateral decubitus position. The 3D data sets were analyzed
offline using the Left Ventricular Analysis System (Siemens Medical
Solutions, Inc., Malvern, PA, USA). Various sequences of the
endocardium were automatically signed and manually revised. The LV
was automatically divided into 17 segments. Diastolic dyssynchrony
index (DDI), as a mechanical dyssynchrony parameter used in the
software, was derived from the standard deviation of the regional
end-diastolic peak volume times of the 17-segment model and
represented the mechanical dyssynchrony of a ventricle.
End-diastolic synchrony with the DDI index was evaluated using the
following equation:
DDI=1N–1∑i=1N(ti–t¯)
Where ti represents regional
relaxation times, and t represents the mean regional
relaxation times.
The images were stored by adopting the P-P interval
in the ECG to acquire a harmonized standard. Then, the sampling
points for every segment were automatically located in the maximum
volume and the DDI was automatically calculated by the
software.
Statistical analysis
Data were expressed as mean ± standard deviation,
data was analyzed using the Chi-square test for qualitative data
and one-way analysis of variance or Wilcoxon rank sum test for
quantitative data, followed by Student-Newman-Keuls multiple range
test for multiple comparisons, and variables were analyzed by
post-hoc tests between the control group and the G+/LVH+ or G+/LVH-
groups. P<0.05 was considered to indicate a statistically
significant difference. All statistical analyses were performed
using the SPSS version 16 for Windows (SPSS, Inc., Chicago, IL,
USA).
Results
Genetic study
Nine mutations in the MyBPC3 gene were
identified in 7/48 cases (15%), with two patients having digenic
mutations in the same gene. Specifically, there are 7 missense
mutations (c.2541C>G/p.Y847X, c.2526C>G/p.Y842X,
c.2527G>T/p.Y843X, c.706A>G/p.S236G, c.772G>A/p.E258K,
c.2971G>A/p.V991M and c.3272A>G/p.D1091G), and 2 synonymous
mutations (c.2118T>C/p.G706G and c.2025G>A/p.G675). Among
them, four mutations were novel while the other five mutations had
been previously published (Table
II). In addition, all the index patients with these novel
mutations have a familial HCM history. Systematic genetic
screenings and echocardiographic characterization of these patients
showed that 18 subjects had MyBPC3 mutations and HCM
phenotype (G+/LVH+ group), and the other 23 subjects had
MyBPC3 mutations only (G+/LVH- group) (Table III). All mutations located in exon
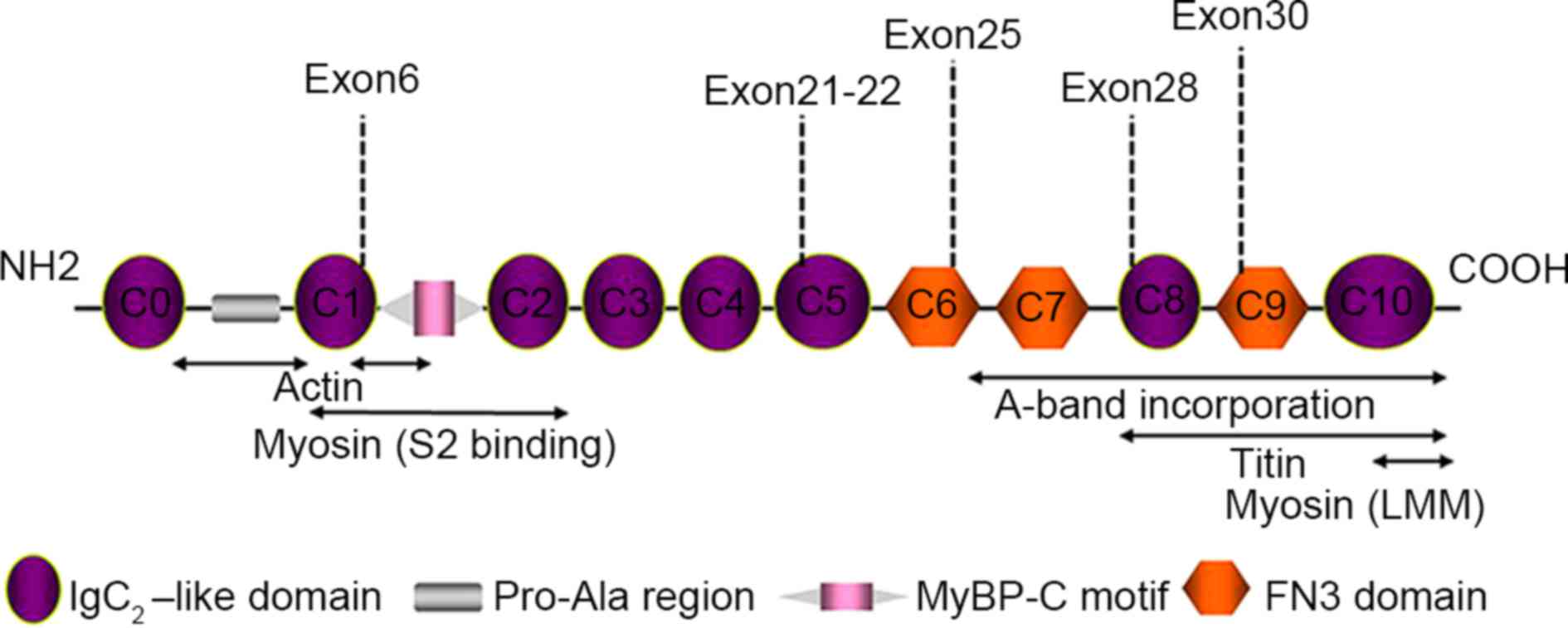
regions were concentrated in 5 myosin domains, C1, C5, C6, C8 and
C9 (Fig. 1).
| Table II.Mutations in the MyBPC3
gene. |
Table II.
Mutations in the MyBPC3
gene.
| Family | Type | Exon | Nucleotide
change | Amino acid
change | Novelty |
|---|
| H104 | Missense | E6 | c.706A>G | p.S236G | Previously
published |
| H31 | Missense | E6 | c.772G>A | p.E258K | Previously
published |
| H09 | Missense | E25 | c.2541C>G | p.Y847X | Previously
published |
| H35 | Missense | E25 | c.2526C>G | p.Y842X | Previously
published |
| H108 | Missense | E25 | c.2527G>T | p.Y843X | Previously
published |
| H104 | Synonymous | E22 | c.2118T>C | p.G706G | Novel |
| H106 | Synonymous | E21 | c.2025G>A | p.G675G | Novel |
| H35 | Missense | E28 | c.2971G>A | p.V991M | Novel |
| H107 | Missense | E30 | c.3272A>G | p.D1091G | Novel |
| Table III.Baseline characteristics of mutations
in the MyBPC3 gene. |
Table III.
Baseline characteristics of mutations
in the MyBPC3 gene.
| Family | Gender | Age (years) | Mutation | Exon | LVH
distribution | G+/LVH+(n=18) | G+/LVH-(n=23) |
|---|
| H104 | M | 55 | c.706A>G
c.2118T>C | E6+E22 | Mid septum and
apical | 2 | 2 |
| H31 | M | 43 | c.772G>A | E6 | Mid septum | 4 | 6 |
| H09 | F | 45 | c.2541C>G | E25 | Mid septum | 4 | 7 |
| H35 | M | 56 | c.2526C>G
c.2971G>A | E25+E28 | Mid septum and
apical | 2 | 2 |
| H108 | M | 55 | c.2527G>T | E25 | Basal-mid
septum | 2 | 2 |
| H106 | F | 35 | c.2025G>A | E21 | Basal-mid
septum | 1 | – |
| H107 | M | 44 | c.3272A>G | E30 | Apical | 3 | 4 |
Clinical characteristics
The clinical variables of subjects enrolled in this
study at the baseline are displayed in Table IV. There was no significant
difference in blood pressure or heart rate among the three groups.
However, by comparing with the other two groups, the G+/LVH+
population was significantly older (P<0.0001) and had more males
than females. All patients were symptomatic at the first
presentation; 90% of them had mild symptoms (functional New York
Heart Association [NYHA] class). Patients with HCM were managed
primarily through medication (β-blockers and calcium channel
blockers). Diastolic function was normal (grade 0) in all controls
and all G+/LVH- subjects. The majority of G+/LVH+ subjects had
grades 1 to 2 diastolic dysfunction and asymmetrical septal
hypertrophy (14/18).
| Table IV.Phenotypic characteristics of
patients. |
Table IV.
Phenotypic characteristics of
patients.
| Characteristic | G+/LVH+ (n=18) | G+/LVH- (n=23) | Controls
(n=30) | P-value |
|---|
| Age, years | 48±12a | 38±10a | 40±11 | <0.001 |
| Men, n (%) | 11 (61%) | 16 (69%) | 19 (63%) | 0.460 |
| Body surface area,
m2 | 1.8±0.2 | 1.6±0.1 | 1.7±0.1 | 0.210 |
| Systolic blood
pressure | 125±16 | 120±10 | 122±9 | 0.090 |
| Diastolic blood
pressure | 74±9 | 70±9 | 73±8 | 0.140 |
| Heart rate,
beats/min | 68±10 | 71±10 | 67±10 | 0.580 |
| NYHA function class
(%) | 2 (11%) | 0 | 0 | <0.001 |
| Dyspnea, n (%) | 11 (61%) | 0 | 0 | <0.001 |
| Angina, n (%) | 7 (39%) | 0 | 0 | <0.001 |
| β-blockers, n
(%) | 10 (56%) | 0 | 0 | <0.001 |
| LVOT obstruction, n
(%) (pressure gradient ≥30 mmHg) | 5 (28%) | 0 | 0 | <0.001 |
| Distribution of
HCM |
|
|
|
|
Asymmetric septum, n (%) | 14 (78%) | 0 | 0 | – |
|
Symmetric septum, n (%) | 0 | 0 | 0 | – |
| Apical
HCM, n (%) | 4 (22%) | 0 | 0 | – |
| Grade of diastolic
function |
|
|
|
| Grade
0 | 0 | 23 | 30 | 0.009 |
| Grade
1 | 6 | 0 | 0 | <0.001 |
| Grade
2 | 8 | 0 | 0 | <0.001 |
| Grade
3 | 4 | 0 | 0 | <0.001 |
Echocardiographic features
The results of the echocardiographic analysis are
displayed in Table V. As expected,
maximal LV wall thickness and interventricular septal thickness
(IVS) were higher in the G+/LVH+ subjects compared with control
(P<0.05) and G+/LVH-subjects. In addition, the left atrial
dimension was significantly higher in the G+/LVH+ subjects compared
with the control. There were no significant differences in LVEDD,
LVESD and LV ejection fraction among the three groups.
| Table V.2-Dimensional echocardiography
characteristics of patients. |
Table V.
2-Dimensional echocardiography
characteristics of patients.
| Characteristic | G+/LVH+ (n=18) | G+/LVH- (n=23) | Controls
(n=30) | P-value |
|---|
| Maximal LV wall
thickness, cm |
2.0±0.3a | 0.9±0.4 | 0.8±0.2 | <0.001 |
| IVS, cm |
1.9±0.5a |
1.0±0.2a | 0.8±0.3 | <0.001 |
| LPW, cm |
0.8±0.2a | 0.7±0.1 | 0.7±0.1 | <0.001 |
| IVS/LPW |
2.4±0.4a |
1.4±0.3a | 1.1±0.2 | <0.001 |
| LVEDD, cm | 4.6±0.8 | 4.7±0.6 | 4.6±0.7 | 0.720 |
| LVESD, cm | 3.2±0.7 | 3.1±0.6 | 3.0±0.5 | 0.680 |
| LV ejection
fraction, % | 62.2±6.8 | 64.0±4.9 | 62.9±5.6 | 0.200 |
| LA, cm |
40.0±3.3a | 35.0±2.1 | 33±2.6 | <0.001 |
| E/A ratio | 1.4±0.7 | 1.3±0.6 | 1.4±0.6 | 0.060 |
| DT, msec |
169.7±49.0a |
157.0±47.0a | 140.0±44.0 | 0.010 |
| Septal Sa,
cm/sec |
8.0±1.5a | 9.8±1.3 | 10.1±1.0 | <0.001 |
| Septal Ea,
cm/sec |
6.2±2.3a |
8.5±1.7a | 11.4±2.1 | <0.001 |
| Septal Aa,
cm/sec |
7.8±2.5a | 11.4±3.1 | 10.6±2.3 | <0.001 |
| Septal E/Ea
ratio |
13.4±3.0a |
6.6±2.4a | 5.2±2.1 | <0.001 |
| Lateral Sa,
cm/sec |
8.4±2.2a | 10.9±2.6 | 12.2±3.1 | <0.001 |
| Lateral Ea,
cm/sec |
9.1±2.3a | 15.9±2.1 | 17.3±1.9 | <0.001 |
| Lateral Aa,
cm/sec | 9.8±3.5 | 10.9±2.8 | 11.6±2.9 | 0.140 |
| Lateral E/Ea
ratio |
8.1±3.6a | 5.7±2.3 | 6.4±2.8 | <0.001 |
| Average Sa,
cm/sec |
8.1±1.8a | 10.4±2.1 | 11.2±2.0 | <0.001 |
| Average Ea,
cm/sec |
7.7±2.9a |
12.6±2.0a | 14.5±1.8 | <0.001 |
| DDI |
9.1±3.4a | 5.1±1.2 | 4.7±1.6 | <0.001 |
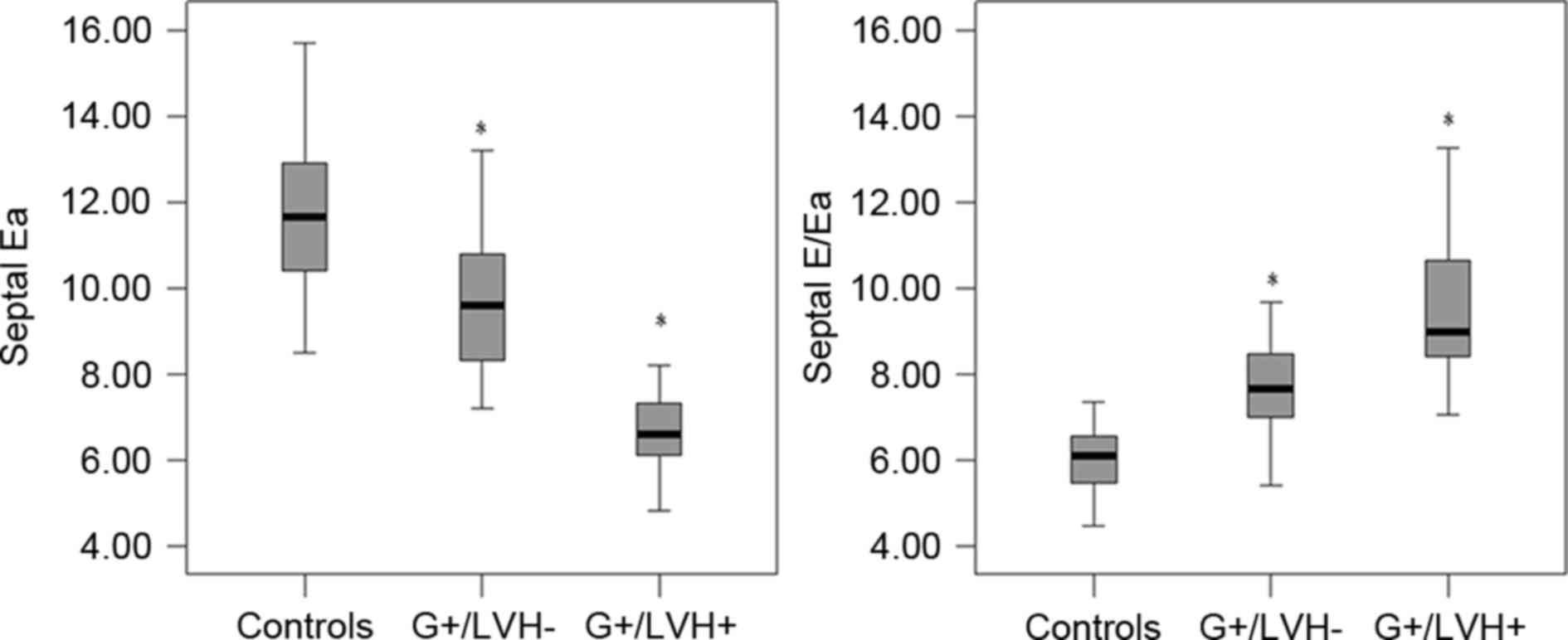
In the G+/LVH+ group, velocity parameters including
septal Sa, Ea and Aa, and lateral Sa and Ea velocities were
significantly lower compared with their counterparts in the control
group (P<0.05). However, there was no significant difference in
lateral Aa velocity between the G+/LVH+ and control groups. In the
G+/LVH- group, only the septal Ea peak velocity (rather than septal
Sa and Aa, and lateral Sa, Ea and Aa velocities) was significantly
lower (P<0.0001) compared with the control group (Fig. 2). Fig.
2 shows the comparison between the G+/LVH- or G+/LVH+ group and
the control. There was no significant difference in septal Sa and
Aa, and lateral Sa, Ea and Aa velocities between the G+/LVH- and
control groups. The DT was significantly increased in the G+/LVH+
and G+/LVH-groups compared with the control group (P<0.0001),
although there was no significant difference in E/A ratios among
the three groups.
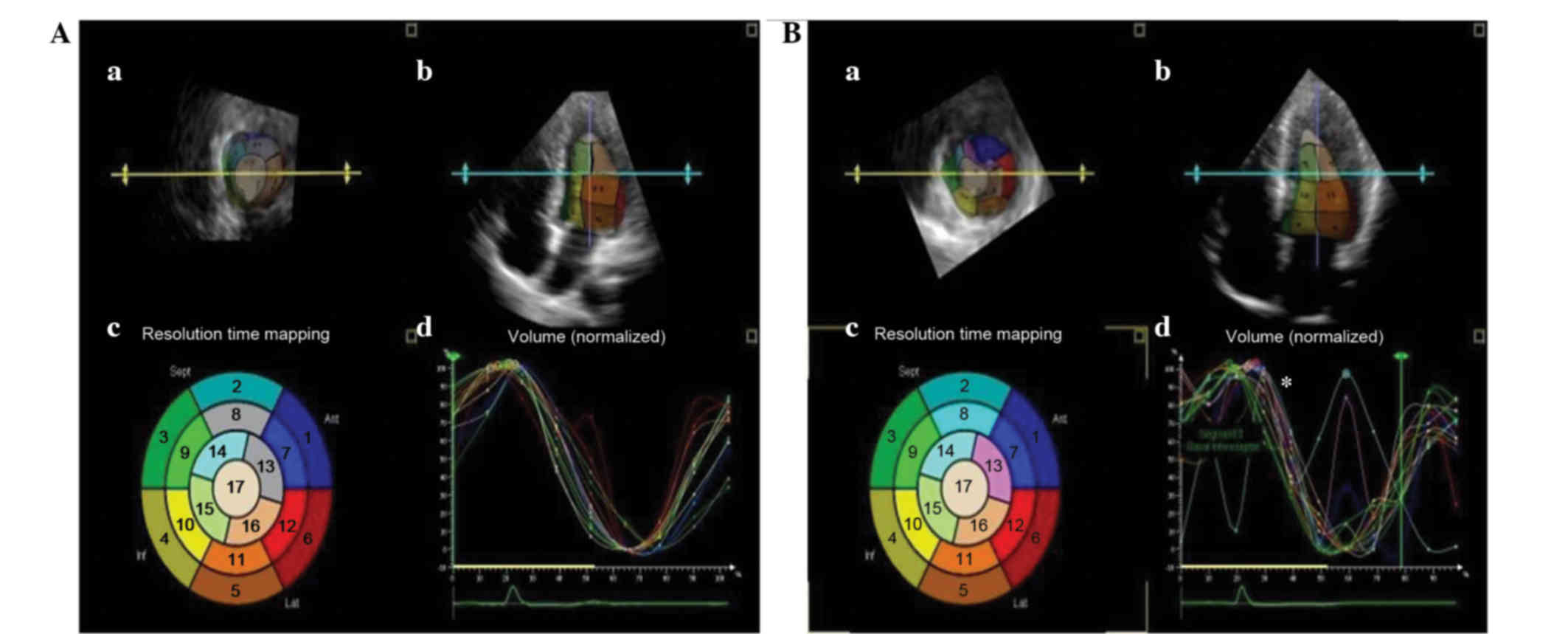
According to the 3DE images, the 17 segmental
curves, divided according to the recommendations of the Joint
Committee of the American Heart Association (17), were smooth and regular for the
controls, whereas the curves for HCM subjects were dyssynchronous
(Fig. 3). A significant increase in
DDI was found in the G+/LVH+ group (P<0.0001). However, no
difference was observed between controls and the G+/LVH- group.
This implies that the diastolic dyssynchrony may be a predominant
cause of the diastolic dysfunction in patients with HCM.
Discussion
HCM is a primary myocardial disorder marked by
genotypic and phenotypic heterogeneity (18). More than 200 different gene mutations
have been reported for HCM, and the disease is typically caused by
single heterozygous mutations in the gene encoding sarcomere
proteins. Mutations in MyBPC3 are the most common type found
in patients with HCM. Identification of specific MyBPC3
mutations may give clinicians insight into the genetic course of
the disease. Various reports have indicated that MyBPC3
mutations are present in 14.5–26% of patients with HCM [18.2% at
the Mayo Clinic, 14.5% at Harvard Medical School, 26% in France,
18% in Germany, 21.7% in Sweden and 24% in Finland (19)]. However, the prevalence of
MyBPC3 mutations in Chinese patients with HCM has, to the
best of our knowledge, not been investigated. The present study
revealed that there is a 15% prevalence of MyBPC3 mutations
in Chinese patients with HCM, which is within the range reported
above. In addition, double MyBPC3 mutations were identified
in two families, which accounts for 3% of the multiple mutation
rate, similar to as previously described (19,20).
These data indicate that MyBPC3 mutations may be the most
common genetic cause of HCM in patients in China.
MyBPC3 is a structural and regulatory protein found
in the sarcomere. The C0 and C1 domains at the N-terminus are
primarily responsible for sarcomere regulation and include a
Pro-Ala-rich region and a myosin binding domain, which is a target
of post-translational modifications. The C5-C10 domains at the
C-terminus participate in binding myosin and titin, and are
necessary for MyBPC3 stability and sarcomere organization (21,22). The
novel mutations identified in the present study are located at C5,
C8 and C9, suggesting that these mutations may affect myocardial
function through changing MyBPC3 protein stability and sarcomere
structure.
Diastolic dysfunction has been documented as the
early sign of HCM preceding LVH in human and animal models
harboring HCM disease causing genetic mutations (23–26).
Previous studies have demonstrated that nonhypertrophied regions of
LV may contribute to diastolic dysfunction in patients with HCM
(27). Diastolic dysfunction was
observed in all patients with HCM with MyBPC3 gene mutations
in the present study, along with significantly increased septal and
lateral E/Ea ratio and significantly increased DDI values. E/Ea
ratio reflects the filling pressure in patients with HCM by TDI,
and the higher ratio reflects a higher severity of symptoms
(28). The DDI value is a mechanical
dyssynchrony parameter in 3DE; the higher the value, the more
severe the diastolic dyssynchrony (29). Furthermore, the present study showed
that regional remodeling of LV may be independent and occur prior
to global LVH. Increased septal E/Ea ratio was observed in the
G+/LVH- group in the absence of lateral E/Ea ratio change, and
increased IVS and LPW ratio in G+/LVH- group was observed, which
suggests local remodeling, consistent with previous studies which
showed histological abnormalities in the absence of significant LVH
(30). In addition, no significant
change in DDI value was observed in the G+/LVH- group, indicating
that regional myocardial function abnormalities were not serious
enough to affect global LV diastolic function. These findings
suggest that regional hypertrophic remodeling proceeds myocardial
dysfunction in patients with MyBPC3 mutations. However,
detailed prospective studies are required to better understand the
chronology of the development of HCM caused by MyBPC3
mutations.
In conclusion, the present study revealed a 15%
prevalence of MyBPC3 gene mutations in the Chinese HCM
population. MyBPC3 gene mutation caused regional LV
hypertrophic remodeling first and further proceeded to global
hypertrophic remodeling and myocardial diastolic dysfunction.
Combined applications of 2DE, TDI and 3DE are a feasible way to
detect early myocardial remodeling and myocardial dysfunction in
patients with genetic predisposition.
References
|
1
|
Elliott P and McKenna WJ: Hypertrophic
cardiomyopathy. Lancet. 363:1881–1891. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Landstrom AP and Ackerman MJ: Mutation
type is not clinically useful in predicting prognosis in
hypertrophic cardiomyopathy. Circulation. 122:2441–2449; discussion
2450. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Watkins H, Rosenzweig A, Hwang DS, Levi T,
McKenna W, Seidman CE and Seidman JG: Characteristics and
prognostic implications of myosin missense mutations in familial
hypertrophic cardiomyopathy. N Engl J Med. 326:1108–1114. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maron BJ, Maron MS and Semsarian C:
Genetics of hypertrophic cardiomyopathy after 20 years: Clinical
perspectives. J Am Coll Cardiol. 60:705–715. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang L, Seidman JG and Seidman CE:
Narrative review: Harnessing molecular genetics for the diagnosis
and management of hypertrophic cardiomyopathy. Ann Intern Med.
152:513–520. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Flavigny J, Robert P, Camelin JC, Schwartz
K, Carrier L and Berrebi-Bertrand I: Biomolecular interactions
between human recombinant beta-MyHC and cMyBP-Cs implicated in
familial hypertrophic cardiomyopathy. Cardiovasc Res. 60:388–396.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kimura A: Molecular etiology and
pathogenesis of hereditary cardiomyopathy. Circ J. 72:(Suppl A).
A-38–A48. 2008. View Article : Google Scholar
|
|
8
|
Ho CY, Lever HM, DeSanctis R, Farver CF,
Seidman JG and Seidman CE: Homozygous mutation in cardiac troponin
T: Implications for hypertrophic cardiomyopathy. Circulation.
102:1950–1955. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Richard P, Charron P, Leclercq C, Ledeuil
C, Carrier L, Dubourg O, Desnos M, Bouhour JB, Schwartz K, Daubert
JC, et al: Homozygotes for a R869G mutation in the beta-myosin
heavy chain gene have a severe form of familial hypertrophic
cardiomyopathy. J Mol Cell Cardiol. 32:1575–1583. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maron BJ, Niimura H, Casey SA, Soper MK,
Wright GB, Seidman JG and Seidman CE: Development of left
ventricular hypertrophy in adults in hypertrophic cardiomyopathy
caused by cardiac myosin-binding protein C gene mutations. J Am
Coll Cardiol. 38:315–321. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wigle ED, Sasson Z, Henderson MA, Ruddy
TD, Fulop J, Rakowski H and Williams WG: Hypertrophic
cardiomyopathy. The importance of the site and the extent of
hypertrophy. A review. Prog Cardiovasc Dis. 28:1–83. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McKenna WJ, Spirito P, Desnos M, Dubourg O
and Komajda M: Experience from clinical genetics in hypertrophic
cardiomyopathy: Proposal for new diagnostic criteria in adult
members of affected families. Heart. 77:130–132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-tie quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spirito P, Bellone P, Harris KM, Bernabo
P, Bruzzi P and Maron BJ: Magnitude of left ventricular hypertrophy
predicts the risk of sudden death in hypertrophic cardiomyopathy. N
Engl J Med. 342:1778–1785. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maron BJ, McKenna WJ, Danielson GK,
Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH III,
Spirito P, Ten Cate FJ and Wigle ED: Task Force on Clinical Expert
Consensus Documents, American College of Cardiology; Committee for
Practice Guidelines, European Society of Cardiology: American
college of cardiology/European society of cardiology clinical
expert consensus document on hypertrophic cardiomyopathy. A report
of the American college of cardiology foundation task force on
clinical expert consensus documents and the European society of
cardiology committee for practice guidelines. J Am Coll Cardiol.
42:1687–1713. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McKenna WJ, Spirito P, Desnos M, Dubourg O
and Komajda M: Experience from clinical genetics in hypertrophic
cardiomyopathy: Proposal for new diagnostic criteria in adult
members of affected families. Heart. 77:130–132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cerqueira MD, Weissman NJ, Dilsizian V,
Jacobs AK, Kaul S, Laskey WK, Pennell DJ, Rumberger JA, Ryan T and
Verani MS: American Heart Association Writing Group on Myocardial
Segmentation and Registration for Cardiac Imaging: Standardized
myocardial segmentation and nomenclature for tomographic imaging of
the heart. A statement for healthcare professionals from the
cardiac Imaging committee of the council on clinical cardiology of
the American Heart Association. Circulation. 105:539–542. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Elliott P and McKenna WJ: Hypertrophic
cardiomyopathy. Lancet. 363:1881–1891. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Van Driest SL, Ommen SR, Tajik AJ, Gersh
BJ and Ackerman MJ: Sarcomeric genotyping in hypertrophic
cardiomyopathy. Mayo Clin Proc. 80:463–469. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Van Driest SL, Vasile VC, Ommen SR, Will
ML, Tajik AJ, Gersh BJ and Ackerman MJ: Myosin binding protein C
mutations and compound heterozygosity in hypertrophic
cardiomyopathy. J Am Coll Cardiol. 44:1903–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Barefield D and Sadayappan S:
Phosphorylation and function of cardiac myosin binding protein-C in
health and disease. J Mol Cell Cardiol. 48:866–875. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oakley CE, Chamoun J, Brown LJ and Hambly
BD: Myosin binding protein-C: Enigmatic regulator of cardiac
contraction. Int J Biochem Cell Biol. 39:2161–2166. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Geisterfer-Lowrance AA, Christe M, Conner
DA, Ingwall JS, Schoen FJ, Seidman CE and Seidman JG: A mouse model
of familial hypertrophic cardiomyopathy. Science. 272:731–734.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Michele DE, Gomez CA, Hong KE, Westfall MV
and Metzger JM: Cardiac dysfunction in hypertrophic cardiomyopathy
mutant tropomyosin mice is trans-gene-dependent,
hypertrophy-independent, and improved by beta-blockade. Circ Res.
91:255–262. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elliott P, Andersson B, Arbustini E,
Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B,
McKenna WJ, et al: Classification of the cardiomyopathies: A
position statement from the European society of cardiology working
group on myocardial and pericardial diseases. Eur Heart J.
29:270–276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Van Driest SL, Vasile VC, Ommen SR, Will
ML, Tajik AJ, Gersh BJ and Ackerman MJ: Myosin binding protein C
mutations and compound heterozygosity in hypertrophic
cardiomyopathy. J Am Coll Cardiol. 44:1903–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Spirito P and Maron BJ: Relation between
extent of left ventricular hypertrophy and diastolic filling
abnormalities in hypertrophic cardiomyopathy. J Am Coll Cardiol.
15:808–813. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nagueh SF, Bachinski LL, Meyer D, Hill R,
Zoghbi WA, Tam JW, Quiñones MA, Roberts R and Marian AJ: Tissue
Doppler imaging consistently detects myocardial abnormalities in
patients with hypertrophic cardiomyopathy and provides a novel
means for an early diagnosis before and independently of
hypertrophy. Circulation. 104:128–130. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marsan NA, Tops LF, Nihoyannopoulos P,
Holman ER and Bax JJ: Real-time three dimensional echocardiography:
Current and future clinical applications. Heart. 95:1881–1890.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McKenna WJ, Stewart JT, Nihoyannopoulos P,
McGinty F and Davies MJ: Hypertrophic cardiomyopathy without
hypertrophy: Two families with myocardial disarray in the absence
of increased myocardial mass. Br Heart J. 63:287–290. 1990.
View Article : Google Scholar : PubMed/NCBI
|