Introduction
Severe pneumonia is an acute respiratory infection
that leads to a large number of mortalities worldwide (1). Klebsiella pneumoniae (K.
pneumoniae) is a common gram-negative bacterium that can cause
destructive infections in human lungs through inflammation and
hemorrhage. Lung infections caused by K. pneumoniae are
typically necrotic (2,3). Pneumonia caused by K. pneumoniae
has a high mortality rate and in alcoholic patients, it is almost
100% (4).
At present, antibiotic therapy is the standard
treatment for pneumonia caused by K. pneumonia; however, a
number of K. pneumoniae strains are resistant to
antibiotics, including ciprofloxacin (5), carbapenem (6) and colistin (7). Due to the ineffectiveness of the
antibiotics currently used, novel therapies that K.
pneumoniae is not resistant to should be developed.
Nuclear factor κB (NF-κB) is an important
transcription factor in chronic inflammatory diseases (8) and can regulate numerous inflammatory
responses. It is activated by certain inflammatory factors
including interleukin 1β (IL-1β), tumor necrosis factor α (TNF-α)
and bacterial lipopolysaccharides (LPS) (9–11). In
unstimulated cells, the activity is inhibited by the inhibitor
protein κB (IκB), of which IκBα has been well studied (12). When cells are stimulated, NF-κB
related signaling pathways are activated by signals including
reactive oxygen species (ROS), IL-1 and TNF-α (13). By contrast, IκB is phosphorylated and
ubiquitinated by IκB kinases, and eventually degraded by
proteasomes (14). Developing NF-κB
targeted therapy to treat severe pneumonia has been a focus of
previous research (15,16). However, to the best of our knowledge,
the effectiveness of IκBα treatment for severe pneumonia has not
yet been investigated.
In the present study, a pneumonia model in rats was
produced by infecting rats with K. pneumonia. These rats
were subsequently treated with IκBα protein. The study aimed to
provide an insight into the development of a novel therapy for
severe pneumonia and improve understanding of the mechanisms of
IκBα and NF-κB.
Materials and methods
Rat model
The K. pneumoniae standard strain
(ATCC700603) was provided by the National Center for Medical
Culture Collections (Beijing, China). K. pneumoniae was
subcultured onto blood agar containing 5% blood (Shanghai Yeasen
Biotechnology Co., Ltd., Shanghai, China) at 37°C for 18–24 h. The
inoculum was subsequently diluted to 10 colony forming units/ml
(CFU/ml) by ddH2O. A total of 40 Sprague-Dawley (SD)
male rats (Shanghai Laboratory Animal Center, Shanghai, China)
weighing 180–260 g and aged 5–8 months were used. They were housed
in stainless steel wire mesh cages in an animal room that was
maintained at 22±2°C and 40–70% relative humidity with air
ventilation 10–15 times/h and natural lighting. Normal feeding and
water were provided. A rat model of pneumonia was established as
previously described (17). Briefly,
the SD rats were anesthetized by intraperitoneal injection of 10%
chloral hydrate (Shanghai Xinfan Biological Technology Co., Ltd.,
Shanghai, China). The trachea of each rat was instilled with 0.3 ml
prepared inoculum. Then, the inoculated rats were held upright for
20 sec. The clinical signs of the inoculated rats including
symptoms of dysphoria, activity situation, body temperature,
breath, hair, somnolence, appetite, response and weight were
observed every day. Age- and gender-matched rats inoculated with
0.3 ml sterile physiological saline solution were used as controls.
After 7 days, the randomly selected model rats and the control rats
were sacrificed by placing the rats in a chamber containing
CO2. Arterial blood was taken and analyzed by a
blood-gas analyzer (NOVA Biomedical, Waltham, MA, USA). Lung lobe
obtained by resection was dried at 80°C for 20 h and the ratio of
the fresh weight to dry weight (F/D) was calculated. The left
principal bronchus was ligated and the alveoli of the right lungs
were lavaged with a bronchoscope. The numbers of white blood cells
(WBCs) and neutrophils (PMNs) in the bronchoalveolar lavage fluid
were assessed. The present study was approved by the ethics
committee of Changhai Hospital (Shanghai, China).
IκBα treatment
The model rats were divided into four groups with 10
rats in each. The four groups of rats were injected with 0.2 ml
physiological saline (PS; control), or 10, 20 or 40 mg/kg IκBα
protein (Abcam, Cambridge, UK) respectively. After 15 consecutive
days, the animals were sacrificed by placing the rats in a chamber
containing CO2. The F/D of superior lobe of right lung
was analyzed. The lavage fluid was collected and the numbers of
WBCs and PMNs were checked using a blood cell counter (Beckman
Coulter, Inc., Brea, CA, USA). Blood-gas analysis of arterial blood
using a blood gas analyzer (Nova Biomedical) was also
performed.
Histological analysis
The lung tissues of rats were fixed with 10% neutral
formalin solution at room temperature for 24 h. Then, the lungs
were dehydrated with increasing concentrations of ethanol,
infiltrated with xylene and embedded in paraffin. The lungs were
sectioned to 5–8 µm and stained with hematoxylin and eosin
(H&E). The sections were then observed with a microscope.
Immunohistochemistry (IHC) assay
Sections with a thickness of 4 µm were mounted onto
slides that were coated with adhesive. Slides were deparaffinized
with xylene, rehydrated with graded concentrations of ethanol, and
incubated with H2O2 at 37°C for 10 min. Then,
the sections were washed with phosphate-buffered saline (PBS) and
heat-mediated antigen retrieval was performed using a microwave.
Following blocking using 5% (v/v) normal goat serum (Shanghai
Yeasen Biotechnology Co., Ltd.) at 37°C for 10 min, sections were
incubated overnight with NF-κB p105/p50 monoclonal antibody
(ab32360; Abcam) at a dilution of 1:1,000. Washing with PBS was
completed three times and the sections were incubated with
biotin-conjugated goat-anti-rabbit immunoglobulin G secondary
antibody (diluted with 3% bovine serum albumin/PBS; ab64257; Abcam)
at 37°C for 30 min. Further washing with PBS of the sections was
completed three times, prior to incubation with horseradish
peroxidase-conjugated streptavidin at 37°C for 30 min. The sections
were then washed again using PBS three times and
3,3′-diaminobenzidine (DAB) was used as chromogenic agent. The
streptavidin-peroxidase IHC kit was purchased from Maxim Biotech,
Inc. (Rockville, MD, USA).
Protein chip detection
A protein chip assay kit (QAR-INF-1; Raybiotech,
Inc., Norcross, GA, USA) was used to detect the expression levels
of inflammatory factors in rats with severe pneumonia. The assay
was performed according to the manufacturer's instructions. In
brief, the chips were dried at room temperature for 2 h and blocked
using block buffer for 1 h. Then, the chips were incubated with
serum for 2 h. Following washing, chips were incubated with
antibody (a biotin-labeled antibody mixture diluted with blocking
buffer, both from the QAR-INF-1 kit) for 2 h and with cyanine 3
(CY3) fluorochrome for another 3 h. The fluorescence intensity was
detected using the GenePix 4000B scanner (Molecular Devices, LLC,
Sunnyvale, CA, USA).
Statistical analysis
The results were presented as mean ± standard
deviation (SD). All statistical analyses were performed using SPSS
12.0 software (SPSS, Inc., Chicago, IL, USA). Comparisons between
groups were conducted using Student's t-test, and P<0.05 was
considered to indicate a statistically significant difference.
Results
Animal model
Following inoculation, the model rats appeared ill
and exhibited symptoms of dysphoria, fervescence, polypnea,
somnolence, decreased appetite, slow response and weight loss.
These symptoms increased in intensity over time. However, no
significant changes were observed in control rats. Compared with
the control rats, the oxyhemoglobin saturation (SaO2)
and arterial partial pressure of oxygen (PaO2) of model
rats decreased significantly (P<0.05; Table I). In addition, partial pressure of
carbon dioxide (PaCO2), carbon dioxide (CO2),
WBC, PMNs and F/D of model rats increased significantly (P<0.05;
Table I). The results indicate that
the production of a severe pneumonia rat model was successful.
 | Table I.Changes in incidence of pneumonia in
model rats and control rats. |
Table I.
Changes in incidence of pneumonia in
model rats and control rats.
| Group | SaO2
(%) | PaO2
(kPa) | PaCO2
(kPa) | CO2
(ml/dl) | WBC
(109/l) | PMN
(109/l) | F/D |
|---|
| Control | 98.47±4.23 | 12.43±1.12 | 6.56±0.32 | 18.87±0.47 | 0.45±0.02 | 0.04±0.00 | 3.42±0.23 |
| Model |
72.36±5.38a |
8.03±0.42a |
9.25±0.58a |
30.21±0.62a |
2.48±0.15a |
2.42±0.25a |
5.46±0.42a |
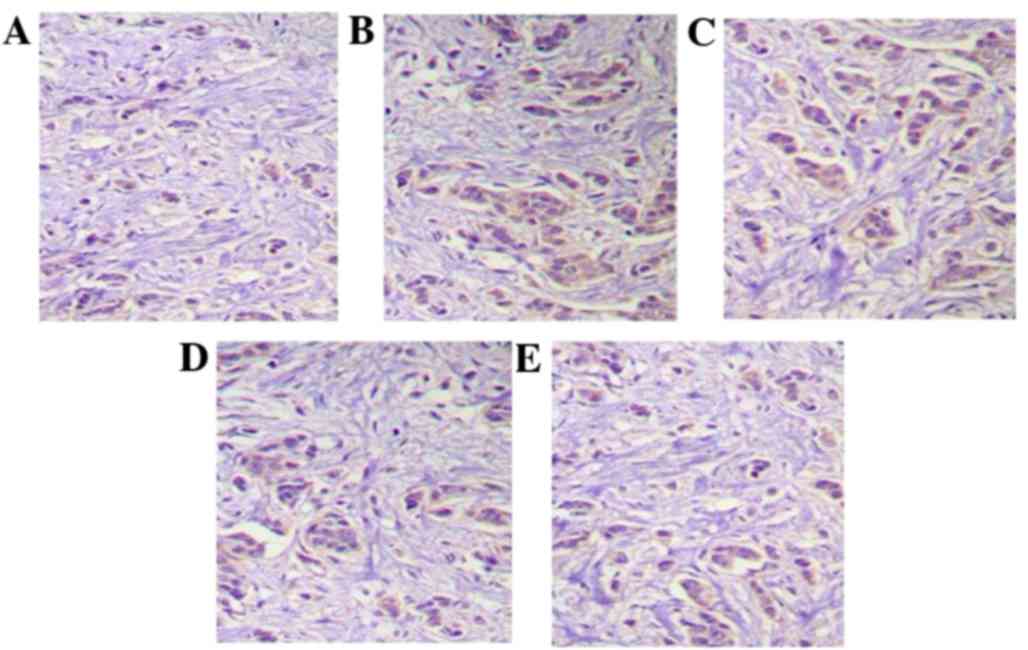
Activity of NF-κB
As presented in Fig.
1, the IHC results demonstrate that in model rats, the
cytoplasm and cell nuclei of NF-κB positive cells were colored
brown. Following middle (20 mg/kg) and high dose (40 mg/kg) IκBα
protein treatment, this brown coloration was significantly
reduced.
Change in the indices of
pneumonia
Table II compares
the level of indices associated with pneumonia in rats treated with
PS or IκBα. The SaO2 and PaO2 of model rats
in the middle and high dose IκBα treatment groups increased
significantly compared with their values in rats treated with PS
(P<0.05), while significant reductions in PaCO2,
CO2, the number of WBC and PMNs and F/D were observed in
rats treated with ≥20 mg/kg IκBα (P<0.05; Table II).
| Table II.Changes in indices of pneumonia in
model rats following different treatments. |
Table II.
Changes in indices of pneumonia in
model rats following different treatments.
| Group | SaO2
(%) | PaO2
(kPa) | PaCO2
(kPa) | CO2
(ml/dl) | WBC
(109/l) | PMN
(109/l) | F/D |
|---|
| Control | 100.25±3.56 | 13.58±0.82 | 5.87±0.42 | 17.56±1.08 | 0.45±0.02 | 0.041±0.002 | 3.12±0.12 |
| Physiological
saline | 70.48±2.24 | 7.93±0.35 | 10.55±0.26 | 32.23±1.78 | 2.48±0.15 | 2.245±0.184 | 6.12±0.23 |
| 10 mg/kg IκBα | 72.89±2.56 | 8.25±0.46 | 9.23±1.58 | 30.68±1.21 | 2.03±0.08 | 2.034±0.076 | 5.87±0.21 |
| 20 mg/kg IκBα |
92.46±4.21a |
12.54±1.12a |
6.86±0.85a |
19.34±2.05a |
1.21±0.01a |
0.123±0.014a |
3.85±0.24a |
| 40 mg/kg IκBα |
96.87±3.54a |
12.26±0.87a |
6.21±0.35a |
18.58±1.02a |
0.87±0.03a |
0.089±0.003a |
3.22±0.38a |
Histopathological analysis
As presented in Fig.
2, the lung of the normal rats was pink, fine and smooth
without exudation and congestion. In addition, pulmonary alveoli
exhibited clear structures, thin walls and clean alveolar spaces.
However, the lung of the pneumonia model rats was dark red with
exudation of blood and purulence. Diffuse pulmonary consolidation
was observed. Following treatment with IκBα, exudation of blood and
purulence of lung in model rats decreased and the group receiving
high dose IκBα treatment demonstrated the least exudation. IκBα
treatment also alleviated the damage of pulmonary alveolus
structure in model rats (Fig.
2).
Expression of inflammatory
factors
As presented in Fig.
3, the expression levels of interleukin 6 (IL-6), TNF-α,
interferon γ (IFN-γ) and monocyte chemoattractant protein-1 (MCP-1)
in PS rats were significantly higher than the control group
(P<0.05). Following treatment with middle and high dose IκBα,
levels of IL-6, TNF-α, IFN-γ and MCP-1 expression were
significantly lower than those indicated for the PS group
(P<0.05, Fig. 3B).
 | Figure 3.Expression of inflammatory factors.
(A) Results of protein chip assay for (a) untreated control rats,
(b) model rats with 0.2 ml physiological saline, (c) 10 mg/kg, (d)
20 mg/kg and (e) 40 mg/kg IκBα protein treatment respectively; (f)
the distribution of the inflammatory factors in each plate. (B)
Relative expression of TNF-α, IL-6, IFN-γ and MCP-1. *P<0.05 vs.
control; #P<0.05 vs. the physiological saline-treated
group. POS, the quality control index of the chip; IκBα, inhibitor
κBα; TNF-α, tumor necrosis factor α; IL, interleukin; IFN-γ,
interferon γ; MCP-1, monocyte chemoattractant protein-1. |
Discussion
In the current study, a rat model for severe
pneumonia was produced by infecting SD rats with K.
pneumoniae. In the model rats, the activity of NF-κB was
significantly higher than that of controls, suggesting that NF-κB
was activated and this activation may facilitate inflammation. By
contrast, when the model rats were treated with IκBα, the activity
of NF-κB was inhibited, and there was a reduction in
PaCO2, CO2, F/D and the numbers of WBC and
PMNs. The expression of inflammatory factors IL-6, TNF-α, IFN-γ and
MCP-1 also decreased. Thus, expression levels of inflammatory
factors may be indicators of NF-κB activation.
One well-accepted hypothesis about NF-κB is that
NF-κB is a response to the prototypical proinflammatory cytokines
TNFα and IL-1, which contribute to host defenses against a number
of pulmonary bacteria (18,19). However, the association between NF-κB
and inflammation cannot be interpreted clearly, as no responses of
IL-1α and IL-1β were found in the current study. Jones et al
(20) have suggested that the
requirement of TNF-α and IL-1 receptors for NF-κB activation in
pneumonia resulting from infection with Streptococcus
pneumoniae was caused by more than gram-negative stimuli
(Escherichia coli). There is a discrepancy in the current
study, as there are indications that TNF-α may be necessary for
NF-κB activation in K. pneumoniae, however this may be due
to the different responses of IL-1 by different gram-negative
bacteria.
Another explanation is that activation of NF-κB may
be related to macrophage activation through certain cytokines (IL-6
and IFN-γ) and chemokines such as MCP-1. IFN-γ can contribute to
the activation and differentiation of macrophages and subsequent
induction of the inflammatory response (21). Chemokine MCP-1 may be a factor that
acts on macrophages and monocytes and contributes to the
recruitment of PMNs (22). In the
current study, similar to TNF-α, expression of IL-6, IFN-γ and
MCP-1 were induced when NF-κB was activated by bacterial infection.
Then, following inhibition of NF-κB by IκBα, the expression of
IL-6, IFN-γ and MCP-1 decreased. PMNs and WBC in pulmonary alveoli
demonstrated a similar trend. These results suggest that the
expression of proinflammatory cytokines and chemokines, and an
alveolar PMN response are important in the resolution of bacteria
infected pneumonia.
In the present study, it is worth noting that no
significant difference was observed in the expression of IL-2
between control and PS treated rats, but IL-2 expression was
induced following treatment with IκBα protein. Leung and Nabel
(23) have reported that the human T
lymphotropic virus-I may induce expression of the IL-2 receptor by
a NF-κB-like factor. It is also indicated that a similar site to κB
exists upstream of IL-2 receptor α. However, the results of the
current study demonstrated that the expression of IL-2 appears to
be dependent on IκBα rather than NF-κB. The mechanisms of NF-κB on
inflammatory factors are complex and remain unclear. Further
studies are needed to evaluate the NF-κB pathway.
In conclusion, the current study demonstrates that
NF-κB inhibition with IκBα protein therapy prevents the development
of pneumonia caused by K. pneumoniae in a rat model. The
therapeutic effect may occur through the responses of several
proinflammatory factors including TNF-α, IL-6, IFN-γ and MCP-1.
However, to clarify the mechanisms of NF-κB and inflammation,
further studies focused on pneumonia infected by other bacteria in
other animal models should be performed.
References
|
1
|
Mathers CD, Boerma T and Ma Fat DM: Global
and regional causes of death. Br Med Bull. 92:7–32. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sahly H and Podschun R: Clinical,
bacteriological, and serological aspects of Klebsiella infections
and their spondylarthropathic sequelae. Clin Diagn Lab Immunol.
4:393–399. 1997.PubMed/NCBI
|
|
3
|
Lin CY, Wheelock AM, Morin D, Baldwin RM,
Lee MG, Taff A, Plopper C, Buckpitt A and Rohde A: Toxicity and
metabolism of methylnaphthalenes: Comparison with naphthalene and
1-nitronaphthalene. Toxicology. 260:16–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jong GM, Hsiue TR, Chen CR, Chang HY and
Chen CW: Rapidly fatal outcome of bacteremic Klebsiella pneumoniae
pneumonia in alcoholics. Chest. 107:214–217. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Paterson DL, Mulazimoglu L, Casellas JM,
Ko WC, Goossens H, Von Gottberg A, Mohapatra S, Trenholme GM,
Klugman KP, McCormack JG and Yu VL: Epidemiology of ciprofloxacin
resistance and its relationship to extended-spectrum beta-lactamase
production in Klebsiella pneumoniae isolates causing bacteremia.
Clin Infect Dis. 30:473–478. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bratu S, Landman D, Haag R, Recco R, Eramo
A, Alam M and Quale J: Rapid spread of carbapenem-resistant
Klebsiella pneumoniae in New York City: A new threat to our
antibiotic armamentarium. Arch Intern Med. 165:1430–1435. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Antoniadou A, Kontopidou F, Poulakou G,
Koratzanis E, Galani I, Papadomichelakis E, Kopterides P, Souli M,
Armaganidis A and Giamarellou H: Colistin-resistant isolates of
Klebsiella pneumoniae emerging in intensive care unit patients:
First report of a multiclonal cluster. J Antimicrob Chemother.
59:786–790. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barnes PJ and Karin M: Nuclear
factor-kappaB: A pivotal transcription factor in chronic
inflammatory diseases. N Engl J Med. 336:1066–1071. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Renard P, Zachary MD, Bougelet C, Mirault
ME, Haegeman G, Remacle J and Raes M: Effects of antioxidant enzyme
modulations on interleukin-1-induced nuclear factor kappa B
activation. Biochem Pharmacol. 53:149–160. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lentsch AB, Czermak BJ, Bless NM and Ward
PA: NF-kappaB activation during IgG immune complex-induced lung
injury: Requirements for TNF-alpha and IL-1beta but not complement.
Am J Pathol. 152:1327–1336. 1998.PubMed/NCBI
|
|
11
|
Beg AA, Finco TS, Nantermet PV and AS Jr
Baldwin: Tumor necrosis factor and interleukin-1 lead to
phosphorylation and loss of I kappa B alpha: A mechanism for
NF-kappa B activation. Mol Cell Biol. 13:3301–3310. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brockman JA, Scherer DC, McKinsey TA, Hall
SM, Qi X, Lee WY and Ballard DW: Coupling of a signal response
domain in I kappa B alpha to multiple pathways for NF-kappa B
activation. Mol Cell Biol. 15:2809–2818. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gloire G, Legrand-Poels S and Piette J:
NF-kappaB activation by reactive oxygen species: Fifteen years
later. Biochem Pharmacol. 72:1493–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Evans SE, Hahn PY, McCann F, Kottom TJ,
Pavlovic ZV and Limper AH: Pneumocystis cell wall beta-glucans
stimulate alveolar epithelial cell chemokine generation through
nuclear factor-kappaB-dependent mechanisms. Am J Respir Cell Mol
Biol. 32:490–497. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abraham E: The dichotomy of inhibiting
nuclear factor kappa-B in pneumonia. Crit Care. 17:1522013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Everhart MB, Han W, Sherrill TP, Arutiunov
M, Polosukhin VV, Burke JR, Sadikot RT, Christman JW, Yull FE and
Blackwell TS: Duration and intensity of NF-kappaB activity
determine the severity of endotoxin-induced acute lung injury. J
Immunol. 176:4995–5005. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bakker-Woudenberg IA, ten Kate MT, Guo L,
Working P and Mouton JW: Improved efficacy of ciprofloxacin
administered in polyethylene glycol-coated liposomes for treatment
of Klebsiella pneumoniae pneumonia in rats. Antimicrob Agents
Chemother. 45:1487–1492. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laichalk LL, Kunkel SL, Strieter RM,
Danforth JM, Bailie MB and Standiford TJ: Tumor necrosis factor
mediates lung antibacterial host defense in murine Klebsiella
pneumonia. Infect Immun. 64:5211–5218. 1996.PubMed/NCBI
|
|
19
|
Ulich TR, Watson LR, Yin SM, Guo KZ, Wang
P, Thang H and del Castillo J: The intratracheal administration of
endotoxin and cytokines. I. Characterization of LPS-induced IL-1
and TNF mRNA expression and the LPS-, IL-1-, and TNF-induced
inflammatory infiltrate. Am J Pathol. 138:1485–1496.
1991.PubMed/NCBI
|
|
20
|
Jones MR, Simms BT, Lupa MM, Kogan MS and
Mizgerd JP: Lung NF-kappaB activation and neutrophil recruitment
require IL-1 and TNF receptor signaling during pneumococcal
pneumonia. J Immunol. 175:7530–7535. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boehm U, Klamp T, Groot M and Howard J:
Cellular responses to interferon-gamma. Annu Rev Immunol.
15:749–795. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maus UA, Waelsch K, Kuziel WA, Delbeck T,
Mack M, Blackwell TS, Christman JW, Schlöndorff D, Seeger W and
Lohmeyer J: Monocytes are potent facilitators of alveolar
neutrophil emigration during lung inflammation: Role of the
CCL2-CCR2 axis. J Immunol. 170:3273–3278. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leung K and Nabel GJ: HTLV-1
transactivator induces interleukin-2 receptor expression through an
NF-kappa B-like factor. Nature. 333:776–778. 1988. View Article : Google Scholar : PubMed/NCBI
|