Introduction
Prostate cancer (PCa) is the most common type of
malignant tumor among men and the annual rate of PCa-related
mortality is increasing rapidly worldwide (1). Treatments for prostate cancer include
active surveillance, surgery, radiation therapy, chemotherapy,
hormonal therapy, high-intensity focused ultrasound, or various
combinations of these strategies. However, therapies for treating
advanced PCa are considered to be largely ineffective (2,3).
Autophagy is an important homeostatic cellular
recycling mechanism responsible for degrading unnecessary or
dysfunctional cellular organelles and proteins in all living cells
(4). Autophagy promotes a cell
survival response and may be triggered by distinct cellular stress,
including nutrient starvation, pathogen-associated molecular
patterns and virus infection (5,6). Cells
that undergo excessive autophagy may undergo cell death in a
non-apoptotic manner (7).
Additionally, the mammalian target of rapamycin (mTOR) signaling
pathways have been reported to be involved in autophagy regulation
in mammalian cells (8,9). Activation of extracellular
signal-regulated kinases 1/2 (ERK1/2) has also been reported to be
associated with autophagic regulation (10).
Sunitinib, which is a multi-targeted receptor
tyrosine kinase (RTK) inhibitor, is approved for the treatment of
advanced kidney cancer, imatinib-resistant gastrointestinal stromal
cancer, pancreas adenocarcinoma and other types of solid-organ
cancer (11–15). This anti-cancer agent has been
described as an efficient therapeutic tool due to its desirable
features in targeting apoptosis and oxidative stress (16,17). In
PCa, preclinical and clinical studies have shown that sunitinib is
able to inhibit PCa angiogenesis and proliferation, inducing
prostate carcinoma cell apoptosis (18,19).
However, while it may be possible that sunitinib may induce
autophagy in PCa, the underlying effect and mechanism of autophagy
in PCa remains unclear.
In the present study, the cytotoxicity and autophagy
induced by sunitinib were explored using PC-3 and LNCaP human PCa
cell lines. The effects of sunitinib on cell proliferation, cell
cycle, autophagy and apoptotic cell death were investigated in PC-3
and LNCaP cell lines. Furthermore, the mechanisms of
sunitinib-induced cell death were examined, specifically ERK1/2
phosphorylation and mTOR signaling.
Materials and methods
Cell culture and materials
PC-3 and LNCaP human PCa cell lines were obtained
from the American Type Culture Collection (Manassas, VA, USA).
Cells were maintained in a 1:1 mixture of Dulbecco's modified
Eagle's medium (DMEM), Ham's F-12 nutrient mixture (HyClone; GE
Healthcare Life Sciences, Logan, UT, USA) and RPMI 1640 medium
supplemented with 10% fetal bovine serum (FBS) (both HyClone; GE
Healthcare Life Sciences), respectively. Sunitinib malate powder
was supplied by Pfizer, Inc., (New York, NY, USA); 3-methyladenine
(3-MA) and U1260 ERK inhibitor were purchased from Sigma-Aldrich
(Merck Millipore, Darmstadt, Germany). All reagents were diluted in
dimethyl sulfoxide (DMSO) and stored at −20°C.
Cell viability assay
Cell viability assays following sunitinib treatment
were performed using a cell counting kit-8 (CCK-8; Dojindo
Molecular Technologies, Inc., Kumamoto, Japan). PC-3 and LNCaP
cells were seeded in 96-well plates (1×104 cells/well)
with culture medium supplemented with 10% FBS and were incubated at
37°C in incubator with an atmosphere of 5% CO2 for 12 h
to allow adherence. Cells were treated with 10 µl culture medium
containing 0, 5, 10 or 20 µmol/l of sunitinib for 24 h. A total of
10 µl CCK-8 was added to the cells, following sunitinib treatment,
and the cells were incubated for a further 2 h at 37°C. A
microplate reader was used to measure the absorbance of each well
at 450 nm, and inhibition rates were calculated as follows:
Viability rate
(%)={[A450(sample)-A450(blank)]/[A450(control)-A450(blank)]}x100.
Cell cycle analysis
Cell cycle distribution was conducted by staining
DNA with propidium iodide (PI; Sigma-Aldrich, Merck Millipore).
Cells were trypsinized, fixed overnight in 70% ice-cold ethanol,
washed twice with PBS, then centrifuged at 223.6 × g. Cells
were incubated with RNase (50 µl at 100 µg/ml) for 10 min, stained
with propidium iodide (200 µl at 50 µg/ml; cat. no. 81845;
Sigma-Aldrich) for 1 h at room temperature. The percentage of cells
in the different phases of the cell cycle were then analyzed with
the FACSAria II flow cytometer using CellQuest7.6.2 software (both
BD Biosciences, San Jose, CA, USA). ModFit v. 3.3.11 (Verity
Software House, Topsham ME, USA) was used for data analysis.
Apoptosis assays
Apoptosis was determined by Annexin V-PE vs.
7-amino-actinomycin D (7-AAD) staining, using a PE Annexin V
Apoptosis Detection kit I (BD Biosciences), according to the
manufacturer's instructions. Cells were analyzed using a
FACSCalibur flow cytometer and CELLQuest software (BD
Biosciences).
Immunofluorescence staining
Cells were seeded on sterile cover slips in 6-well
tissue culture plates at a concentration of 1×105
cell/ml in a volume of 0.6 ml. Following treatment with either DMSO
(control) or sunitinib (5, 10 or 20 µmol/l) for 24 h, cells were
fixed onto slides with 4 % (v/v) paraformaldehyde for 20 min,
blocked with 5% bovine serum albumin for 30 min at room temperature
and incubated with microtubule associated protein 1A/1B-light chain
3 (LC3) antibody (cat. no. sc292354; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA; dilution 1:200) overnight at 4°C.
Subsequently, the treated cells were washed three times with PBS
and incubated at room temperature with AlexaFluor 488-conjugated
anti-rabbit secondary antibody (cat. no. A-11008; Thermo Fisher
Scientific, Inc., Waltham, MA, USA; dilution 1:1,000) for 1 h.
Fluorescence was measured using a confocal microscope (Leica
Microsystems, Inc., Buffalo Grove, IL, USA; ×630 magnification).
Quantitation of the LC3 puncta was performed by counting 10 cells
manually for each sample.
Western blot analysis
Cells were washed twice with ice-cold PBS and lysed
in lysis buffer (1% NP-40, 5 mM NaPPi, 150 mM NaCl, 20 mM Tris HCL
(pH 7.5), 5 mM Na3VO4, 1 mM PMSF and 10 µg/ml
leupeptin). Samples were vortexed briefly, incubated in lysis
buffer for 30 min on ice and centrifuged at 15,000 × g for
15 min. Protein concentration was determined by the Bradford
method. Equal amounts of samples containing total protein (20 µg)
were separated using 10–15% SDS-PAGE and transferred onto
polyvinylidene fluoride membranes (cat. no. IPVH00010; Merck KGaA,
Darmstadt, Germany). Membranes were blocked in 5% non-fat milk for
1 h at room temperature, incubated with the below indicated primary
antibodies in nonfat milk overnight at 4°C, then washed with
PBS/0.1% Tween 20 for 1 h. Membranes were then incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG (cat. no.
111-035-003; Jackson Immunoresearch Labs, Inc., West Grove, PA,
USA; dilution, 1:20,000) or rabbit goat anti-mouse IgG (cat. no.
115-035-003; Jackson Immunoresearch Labs, Inc.; dilution 1:20,000)
for 1 h. Subsequently, membranes were washed with PBS/0.1% Tween-20
for 40 min and detected with enhanced chemiluminescence. Primary
antibodies used in western blotting, according to the
manufacturer's instructions, were: Anti-LC3, anti-sequestosome-1
(SQSTM1/p62), anti-mTOR, anti-p-mTOR (Ser2481), anti-p-p70S6K
(Thr389) (Cell Signaling Technology, Inc., Danvers, MA, USA),
β-actin, anti-p-ERK1/2 (Thr202/Tyr204), anti-ERK1/2 (Santa Cruz
Biotechnology) and anti-cleaved-caspase3 (Beyotime Institute of
Biotechnology, Jiangsu, China). Bands were revealed using an
enhanced chemiluminescence reagent (ECL) in an ECL Plus kit
(Beyotime Institute of Biotechnology, Jiangsu, China) and recorded
on X-ray films (Fujifilm Life Science, Tokyo, Japan).
MTT assay
Cell viability was measured by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (cat. no. 11465007001; Roche Diagnostics, Basel,
Switzerland). Cells were plated at 5,000 cells/well in 96-well
plates, in triplicate. Cells were allowed to adhere overnight, and
medium containing the test drug or control media was added. After
incubation for 48 h at 37°C in 5% CO2, the
drug-containing medium was removed and replaced by 100 µl fresh
medium with 0.5 mg/ml MTT solution. After incubation for 4 h, the
medium with MTT was removed and 100 µl solubilization solution was
added to each well. The plates were then gently agitated until the
color reaction was uniform, and the OD570 (optical
density at a wavelength of 570 nm) was determined using a
microplate reader (Wellscan MK3, Labsystems Diagnostics, Vantaa,
Finland). Media-only treated cells served as the indicator of 100%
cell viability. Viability rate
(%)={[A570(sample)-A570(blank)]/[A570(control)-A570(blank)]}x100.
Statistical analysis
All results presented were confirmed in at least
three independent experiments. Data were presented as mean ±
standard deviation. Statistical analysis was performed using
one-way analysis of variance with Bonferroni's post-hoc tests for
multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
Sunitinib inhibits the growth of PC-3
and LNCaP cells in vitro
To ascertain the inhibitory effect of sunitinib on
the growth of PCa cells, cell viability assays were performed using
PC-3 and LNCaP cells, either in the presence of sunitinib at
different concentrations (5, 10 or 20 µmol/l) or in the absence of
sunitinib and treated instead with DMSO (control) for 24 h.
Significant inhibitory effects on cell growth in PC-3 and LNCaP
cells were observed in a dose-dependent manner (Fig. 1A and B, respectively), which may
suggest that sunitinib possesses a potent cytotoxic activity
against both human PCa cell lines.
Sunitinib induces G1-phase arrest in
PC-3 and LNCaP cells
Cell viability assays revealed that sunitinib
treatment significantly inhibited the proliferation of PC-3 and
LNCaP cells. Subsequently, whether the inhibition effect on cell
proliferation was related to cell cycle progression was detected.
Flow cytometric analysis indicated that 10 µM sunitinib treatment
for 48 h resulted in the accumulation of cells in the G1 phase in
PC-3 cells, (76.36±4.78%) compared with the control (51.82±7.02%),
and in LNCaP cells (65.83±7.99%) compared with the control
(53.32±6.08%) (Fig. 2). Furthermore,
the populations of PC-3 and LNCaP cells in G2/M phase decreased
significantly (P=0.037 and 0.032 for PC-3 and LNCaP, respectively),
from 19.42±2.49% and 20.65±1.85% in the control groups to
(8.58±1.03%) and (12.11±1.17%) after sunitinib treatment (Fig. 2). These results indicated that
sunitinib induced G1-phase arrest in PC-3 and LNCaP cells.
Sunitinib induces autophagy in PCa
cell lines
Sunitinib, which is a multi-targeted tyrosine kinase
inhibitor, exhibits anti-angiogenic and anti-tumor activity and has
been approved by a large body of research (20,21). It
was speculated that sunitinib may also induce autophagy in PCa
cells. Western blot analysis of SQSTM1/p62 and LC3 and subsequent
immunofluorescence staining of LC3 puncta was performed to evaluate
autophagy. PC-3 and LNCaP cells were treated for 24 h with various
concentrations (5, 10 or 20 µmol/l) of sunitinib or DMSO (control),
respectively. Cells were subjected to immunofluorescence staining
and dose-dependent formation of LC3 fluorescence dots was observed.
Limited numbers of LC3 punctas were observed in control groups
(DMSO) in both cell lines. However, following sunitinib treatment,
the LC3 puncta numbers increased significantly (when comparing the
5, 10 or 20 µmol/l groups with the control, P<0.001 for PC-3,
and P<0.002 for LNCaP), corresponding with increasing
concentrations of sunitinib (Fig. 3A and
B).
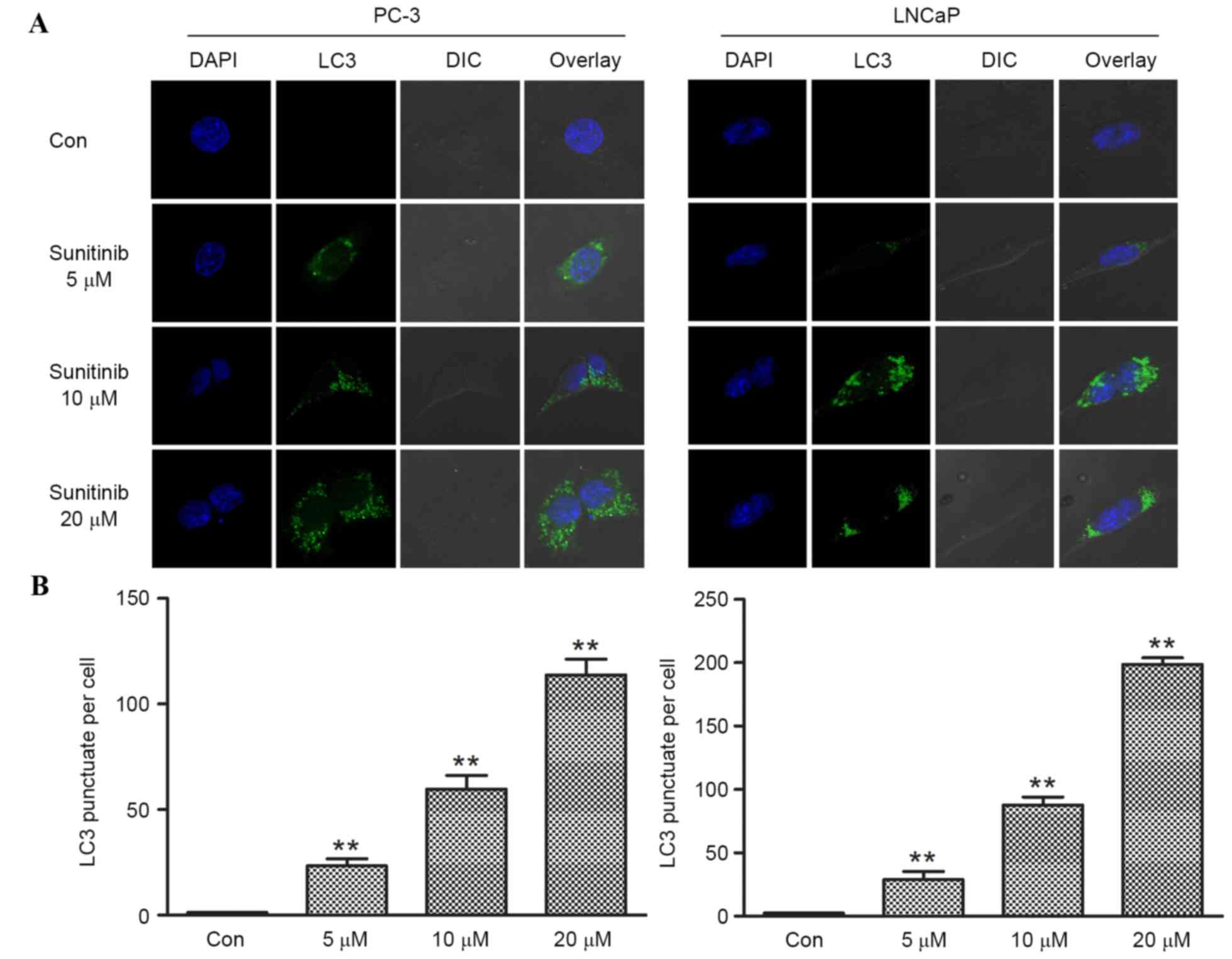 | Figure 3.Effect of sunitinib on the formation
of LC3 puncta in PC-3 and LNCaP cells. (A) Fluorescence images
obtained from sunitinib-treated and control groups of prostate
cancer cells, PC-3 and LNCaP. Cells treated with DMSO were set as
control groups, the numbers of LC3 puncta per cell were counted
manually. The number of LC3 puncta (green fluorescent) per cell in
sunitinib-treated groups increased in a dose dependent manner
(magnification, ×630). (B) Limited numbers of punctas were located
in control group PC-3 and LNCaP cells. The numbers of LC3 puncta
per cell in PC-3 cells of 5, 10 and 20 µM sunitinib-treated groups
were 23.29±3.36, 59.74±6.48 and 113.71±7.52, respectively. The
numbers of LC3 puncta per cell in LNCaP cells of 5, 10 and 20 µM
sunitinib-treated groups were 28.90±6.34, 87.56±6.45 and
198.56±5.33, respectively. Data are presented as means ± standard
deviation (n=4). *P<0.05 and **P<0.01, vs. Con. DSMO,
dimethyl sulfoxide. LC3, microtubule associated protein 1A/1B-light
chain 3; Con, control; DAPI, 4′,6-diamidino-2-phenylindole; DIC,
differential interference contrast. |
A hallmark of autophagy is the enhanced conversion
of microtubule-associated protein 1 light chain 3 (LC3-I) to its
faster-migrating form LC3-II, which is closely associated with the
membrane of autophagosomes. Degradation of SQSTM1/p62 is used as a
marker of autophagic flux (22). p62
is incorporated into autophagosomes and degraded in autolysosomes.
Compared with the control groups, significantly increased
expression levels of LC3-II (when comparing the 5, 10 and 20 µmol/l
groups with the control, P=0.017, <0.001 and <0.001 for PC-3,
and P=0.028, <0.001 and <0.001 for LNCaP, respectively),
accompanied by decreased expression levels of SQSTM1/p62, were
observed in dose-dependent manners in PC-3 and LNCaP cells exposed
to sunitinib for 24 h (Fig. 4A-C).
These findings indicated that sunitinib may have induced autophagy
in the human PCa cell lines.
Regulation of phosphorylated ERK1/2
and mTOR signaling contributes to sunitinib-induced autophagy in
PC-3 and LNCaP cells
Previous studies have confirmed that activation of
AKT/mTOR pathway negatively regulates autophagy (23,24). In
order to assay the direct effect of sunitinib on the activation or
inhibition of ERK and mTOR signaling, western blot analysis was
performed to explore the role of specific components involved in
autophagy flux regulation by observing the expression levels of
total ERK1/2, mTOR, phosphorylated ERK1/2, mTOR and p70S6K.
Treatment with sunitinib at different concentrations for 24 h
induced a significant upregulation of phosphorylated ERK1/2 (when
comparing the 5, 10 or 20 µmol/l groups with the control, P=0.050,
0.003 and <0.001 for PC-3, and P=0.044, 0.048 and <0.001 for
LNCaP, respectively) in a dose-dependent manner for both PCa cell
lines (Fig. 5). Furthermore, the
expression levels of mTOR and its substrate, p70S6K, and their
phosphorylated forms, were investigated to reveal whether the mTOR
pathway may be responsible for sunitinib-induced autophagy. The
results revealed that 10 and 20 µmol/l sunitinib treatment
significantly inhibited the phosphorylation of mTOR (P=0.008 and
0.002 for PC-3, and P=0.005 and 0.001 for LNCaP, respectively) in
both cell lines. Simultaneously, the phosphorylation of p70S6K was
also inhibited (when comparing the 5, 10 or 20 µmol/l groups with
the control, P=0.003, <0.001 and <0.001 for PC-3, and
P=0.010, 0.008 and <0.001 for LNCaP, respectively) (Fig. 5).
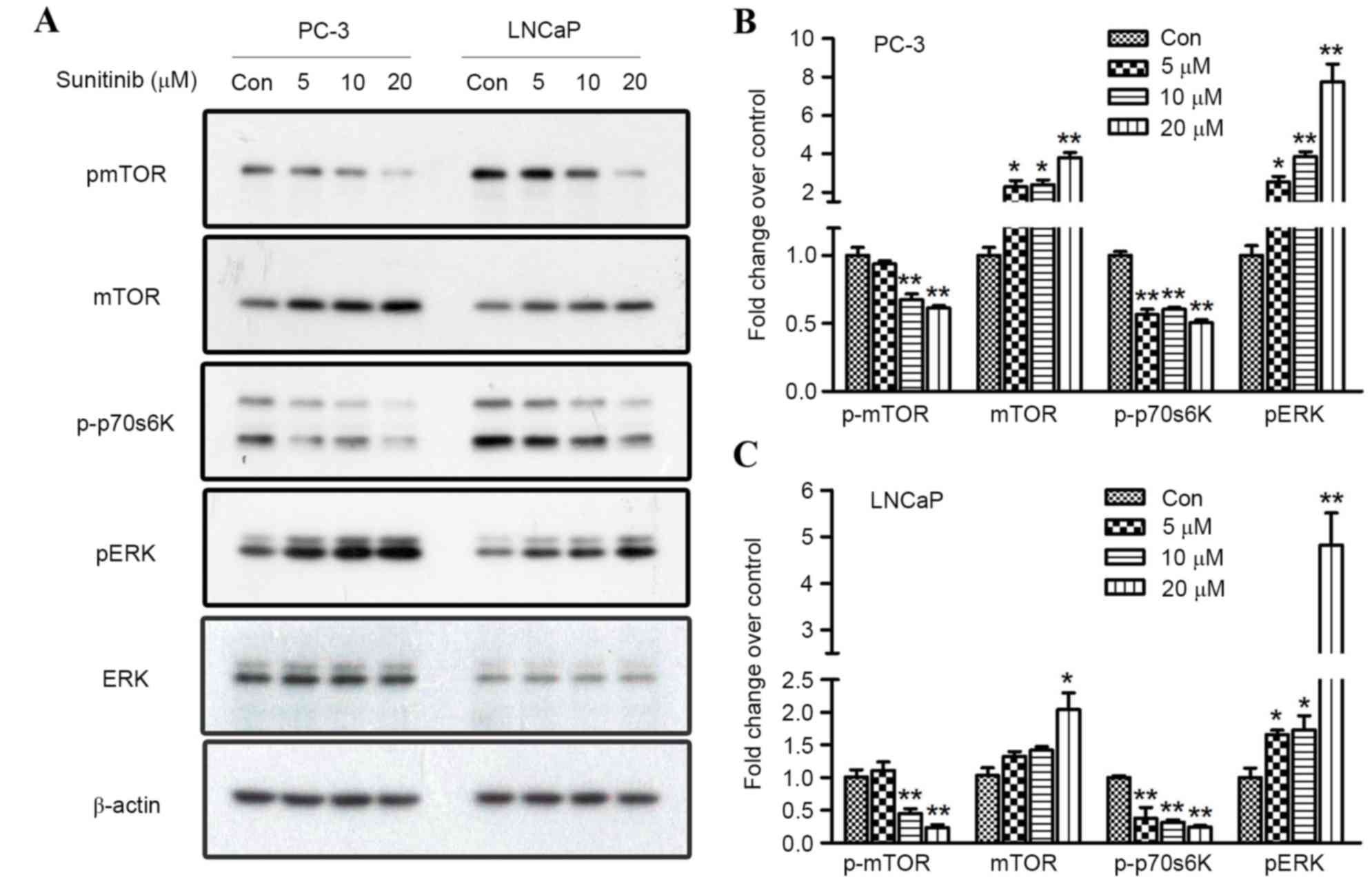 | Figure 5.Effect of sunitinib on mTOR and ERK
signaling in PC-3 and LNCaP cells. (A) Sunitinib treatment
suppressed the phosphorylated levels of mTOR and p70S6K and
activated the phosphorylated level of ERK1/2, as demonstrated by
western blotting. Fold changes in p-mTOR, mTOR, p-p70S6K and p-ERK
proteins in 5, 10, or 20 µM sunitinib-treated groups and control
groups in (B) PC-3 cells and (C) LNCaP cells. Results were
determined by three independent experiments and normalized by the
total protein level. Data are presented as mean ± standard
deviation. *P<0.05 and **P<0.01, vs. Con. mTOR, mechanistic
target of rapamycin; p70S6K, ribosomal protein S6 kinase beta-1;
ERK 1/2, extracellular signal-regulated kinases 1/2; Con,
control. |
U0126 is a highly selective inhibitor of both MEK1
and MEK2, which are types of MAPK/ERK kinase (25). U0126 is able to block the ERK pathway
(26); therefore, U0126 (10 µM) was
used to inhibit the phosphorylation of ERK1/2 in the present study.
LC3II/I ratio was significantly increased when PC-3 and LNCaP cells
were treated with 10 µM sunitinib; however, a decrease in the
LC3II/I ratio was observed when comparing the U1026+sunitinib group
with the sunitinib group in PC-3 and LNCaP cells (P=0.008 and
<0.001, respectively). In addition, quantitation analysis
revealed that when phosphorylation levels of ERK were inhibited, no
significant increase of LC3II/I ratio was found after sunitinib
treatment when compared with the control (Fig. 6). Overall, these results suggest that
ERK1/2 and mTOR activation stimulates sunitinib-induced autophagy
in PC-3 and LNCaP cells.
Autophagy and apoptosis have roles in
sunitinib-mediated cell death in PC-3 and LNCaP cells
To investigate whether apoptosis or autophagy is
involved in the growth inhibitory effect of sunitinib, 3-MA (5 µM)
was administered as a pre-treatment to inhibit sunitinib-induced
autophagy and flow cytometry using Annexin V-PE and 7-AAD staining
was performed, following 10 µM sunitinib treatment for 48 h in both
cell lines. Cleaved caspase-3 protein expression levels were also
determined using western blot analysis.
Significant reductions in the number of viable cells
(Annexin V-PE−/7-AAD−) after treatment with
sunitinib only (sunitinib groups) were observed in PC-3 and LNCaP
cell lines when compared with controls (P=0.007 and 0.038,
respectively), accompanied by significant increases of the number
of apoptotic cells (P=0.001 and 0.017, respectively) (Annexin
V-PE+/7-AAD−, early apoptosis; Annexin
V-PE+/ 7-AAD+, late apoptosis) (Fig. 7A and B). However, there was no
significant difference observed between the 3-MA and control groups
for apoptosis induction. However, when compared with sunitinib +
3-MA groups and 3-MA groups, combined sunitinib and 3-MA treatment
resulted in significantly increased apoptotic cell death (P=0.003
and 0.010 for PC-3 and LNCaP cells, respectively), with a higher
percentage of 7-AAD+ cells. These results indicated that
inhibition of autophagy by 3-MA enhanced sunitinib-induced
apoptosis. Western blot analysis was carried out to detect the
expression of cleaved caspase-3, a hallmark of apoptotic cell death
which preceded the previously observed changes in nuclear
morphology (27). Sunitinib groups
or sunitinib + 3-MA groups exhibited increased expression levels of
cleaved caspase-3 when compared with the control or 3-MA groups.
Furthermore, inhibition of autophagy by 3-MA significantly enhanced
the expression levels of cleaved caspase-3 induced by sunitinib,
when comparing the sunitinib groups with the sunitinib + 3-MA
groups (P=0.019 and 0.038 for PC-3 and LNCaP, respectively)
(Fig. 7C).
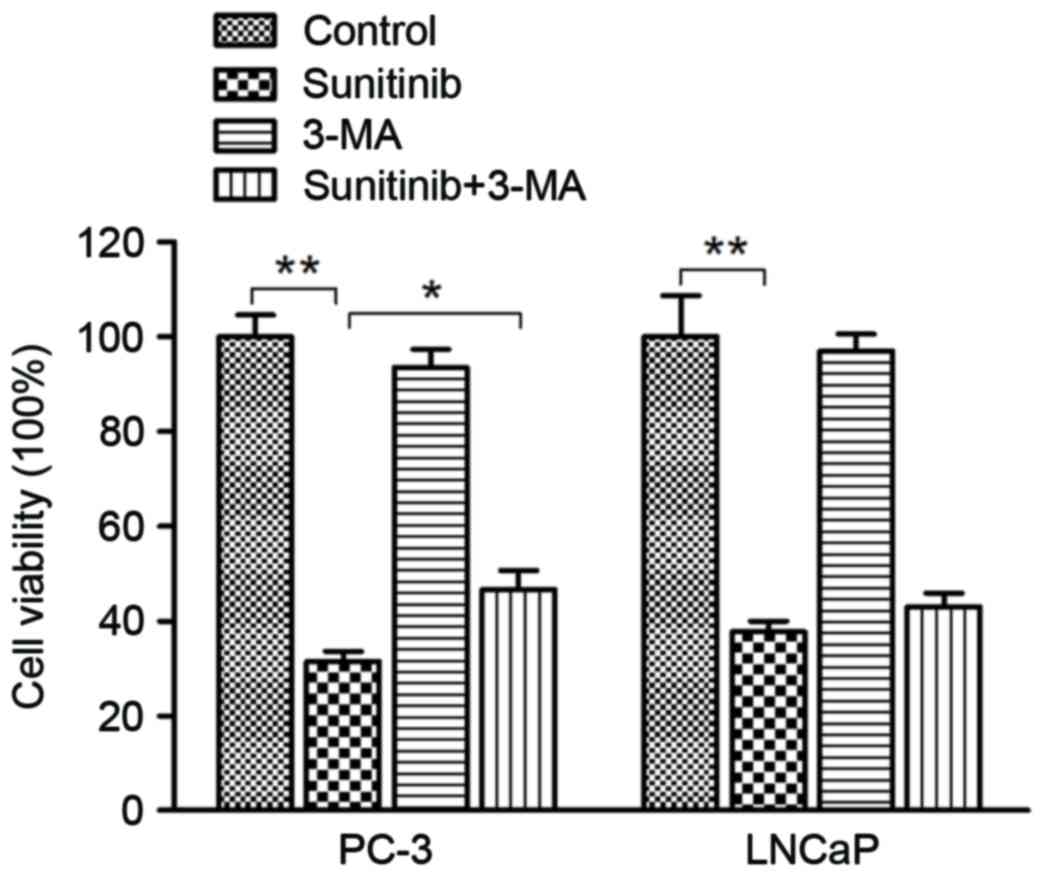
To further evaluate the interplay between apoptosis
and autophagy and their effect on sunitinib cytotoxicity in PCa
cells, a cell viability assay by MTT was performed. Following 48 h
of sunitinib treatment, inhibition of autophagy by 5 µM 3-MA
pre-treatment lead to a significant increase in the rate of cell
viability when comparing the sunitinib group with the sunitinib +
3-MA group in PC-3 cells (P=0.40) (Fig.
8). A similar observation in the increased rate of cell
viability was exhibited in LNCaP cells, although, no statistical
significance was indicated. In consideration of the flow cytometry
assay results, we conclude that sunitinib-triggered cytotoxicity
was mediated by autophagy and apoptotic cell death induction in
PC-3 and LNCaP cells.
Discussion
Previous studies have advanced understanding of the
role of vascular endothelial growth factor (VEGF), which has led to
the addition of several agents to the therapeutic landscape for
various tumor types. Among these antiangiogenic agents, sunitinib
is a tyrosine kinase inhibitor that targets VEGF receptors 1, 2 and
3 and has been approved for the treatment of metastatic renal
carcinoma (28) and
gastro-intestinal stromal tumor (29).
In accordance with our results, several studies have
observed a significant inhibitory and cytotoxicity effect on PCa
cell lines via the regulation of hypoxia and angiogenesis (18,30).
These findings suggest a possible antitumor effect of sunitinib in
PCa. However, there is a lack of studies on the antitumor effect
and mechanism of action of sunitinib in PCa.
In the present study, sunitinib was revealed to
inhibit the viability of PC-3 and LNCaP cells in a dose-dependent
manner. Our findings are in accordance with the results of two
previous studies, concerning anti-tumor and radiosensitivity of
sunitinib on PC-3 and DU145 cells in vitro (31) and the preventative effects in the
course from non-castration to castration of LNCaP xenograft
prostate (19). In addition, cell
cycle analysis in the present study revealed that, following 10 µM
of sunitinib treatment for 24 h, the number of PC-3 and LNCaP cells
in S and G2/M phases was decreased, whereas the number of cells in
G1 phase was increased, when compared with the controls. Similar
results were reported when colonic stromal fibroblasts were treated
with sunitinib mesylate (32).
Moreover, Di Desidero et al (33) identified that sunitinib induced a
concentration-dependent inhibition of the cyclin-D1 gene and
protein expression, in HMVEC-d, 8305C and FB3 cells,
respectively.
Autophagy is a highly conserved, homeostatic process
by which intracellular constituents are delivered to lysosomes for
degradation. It is activated in response to various environmental
stresses that have been documented in human tumor cells, following
treatment with chemotherapeutic drugs (34,35).
Furthermore, a previous study has indicated that excessive
activation of autophagy may lead to another form of programmed cell
death in a non-apoptotic manner (36). LC-3 is considered to be a strong
marker of autophagy. Furthermore, the conversion of LC3-I to LC3-II
and formation of LC3 puncta usually demonstrates the activation of
autophagy (37). In addition, p62
degraded alongside autophagosomal contents, resulting in decreased
p62 levels (38). In the present
study, sunitinib treatment was revealed to promote autophagy in
PC-3 and LNCaP cells in dose-dependent manner, as indicated by the
increases in the LC3-II/LC3-I ratio and membrane-bound lapidated
form of LC3, and the degradation of p62 protein observed. Ikeda
et al (39) reported that
sunitinib induced autophagy in rat pheochromocytoma PC12 cells and
that this was dependent on the suppression of mTORC1 signaling and
the formation of ULK1/2-Atg13-FIP200 complexes. In addition,
another study discovered that sunitinib treatment for 24 h triggers
incomplete autophagy, impaired cathepsin B activation and
stimulated lysosomal-dependent necrosis in bladder cancer cells
(40). To the best of our knowledge,
the present study is the first to propose that autophagy in PCa
cell lines may be stimulated by sunitinib.
mTOR and ERK1/2 are two major pathways that regulate
autophagy induced by nutrient starvation. Cadmium (41) or TNFα treatment (42) have been clearly associated with ERK
activation and autophagic programmed cell death; moreover, direct
ERK activation by the overexpression of active MEK may promote
autophagy without any other stimulus (43). The modifications exhibited by
phosphorylated ERK (p-ERK) after sunitinib treatment are
conflicting, according to previous reports (44,45).
Sunitinib treatment has been revealed to exhibit increased levels
of p-ERK in bladder cancer cells (J82 and 5637) (40) and adrenocortical carcinoma (SW13)
cells (46), whereas p-ERK levels
were suppressed in human colonic stromal fibroblasts (32) and papillary thyroid cancer cells
(44). The present study revealed
that sunitinib significantly promoted ERK1/2 phosphorylation and
suppressed mTOR/p70S6K phosphorylation, which was consistent with
the differences of LC3-II/LC3-I ratio and p62 expression observed.
Furthermore, the highly selective MEK1/2 inhibitor, U0126, markedly
reversed the induction of autophagy by sunitinib when compared with
the sunitinib and U0126 combination group and the sunitinib group,
which suggested that ERK signaling may have a role in
sunitinib-induced autophagy. However, Diaz et al (31) reported that sunitinib promoted a
decrease in the expression level of p-ERK in PC-3 cells after
treatment. Considering that the maximum concentration of sunitinib
used by Diaz et al (31)
employed in that study was 5 µM, which is much lower than that of
our study, both results may be reasonable and further research is
required. It has been established that mTORC1 may suppress
autophagy by promoting phosphorylation and inactivation of proteins
involved in autophagosome formation (47,48). The
role of the mTOR substrate, p70S6Kinase 1 (S6K1), in autophagy
remains controversial (49).
However, one study has indicated that active S6K1 may decrease the
level of LC3 processing and foci formation by autophagosomal
vacuoles in cells treated with sulforaphane, and diminished levels
of S6K1 or a lack of S6 kinases resulted in the accumulation of
autophagosomes in PC-3 cells (50).
This is consistent with the variation trend of p-mTOR and p-p70S6K
demonstrated in the present study, which suggested that mTOR
signaling is also involved in the regulation of sunitinib-induced
autophagy. Overall, the present data revealed that sunitinib may
promote autophagy in PCa cells through the activation of ERK1/2 and
the inhibition of mTOR.
Autophagy, which is described as a ‘double-edged
sword’, has a key role in tumorigenesis, progression and
oncotherapy. On the one hand, autophagy manifests as a survival
pathway that allows tumor cells to live through various severe
environments (51); whereas,
autophagy has also been revealed to contribute to cell death, which
was previously confirmed in cancer cells undergoing antineoplastic
therapy (52). To establish the role
of autophagy in sunitinib-induced cell death, 3-MA (5 µM) was used
as a pre-treatment to block autophagy and subsequent flow cytometry
and cell proliferation assays were employed to determine apoptosis
and cell viability. These analyses revealed that sunitinib
treatment induced an increase in the rate of cellular apoptosis,
while inhibition of autophagy by 3-MA led to a remarkable
enhancement in sunitinib-induced apoptosis. Furthermore, the
expression of cleaved caspase-3 protein, a cysteine protease
involved in the ‘execution’ phase of cellular apoptosis and a key
regulator of tumor repopulation promoting generated from the dying
cells (53), was detected by western
blotting. Western blot analysis showed a similar changing trend in
accordance with the change of apoptosis rate: Sunitinib only
treatment induced significant increases in the number of apoptotic
cells, while a further enhancement of the apoptotic rate was
detected after the inhibition of autophagy by 3-MA when compared
sunitinib + 3-MA group with sunitinib group. Coincidentally,
inhibition of autophagy by 3-MA significantly enhanced the
expression levels of cleaved caspase-3 induced by sunitinib, when
comparing sunitinib groups with sunitinib + 3-MA groups. These
findings suggest that, besides promoting autophagy, sunitinib may
also generate apoptosis in a caspase-3-dependent manner in PC-3 and
LNCaP cells. Additionally, 3-MA (5 µM) was used to inhibit
autophagy and cell viability following 48 h of 10 µM sunitinib
treatment was determined by MTT. Unexpectedly, the cell death rate
of PC-3 cells significantly decreased when treated with sunitinib
and 3-MA pre-treatment; despite this, the inhibition of autophagy
lead to a significant enhancement of sunitinib-induced apoptosis.
Similar observations were identified in LNCaP cells; however, no
statistical significance was detected. Collectively, these findings
indicated that apoptosis and autophagic cell death have key roles
in sunitinib-induced cytotoxicity in PCa cells. While relatively
few reports focus on the induction of autophagy by sunitinib
(39,40), the present results suggest that
autophagy may be an important component in the cytotoxicity
resulting from sunitinib treatment in PCa cells.
Phase II trials of single-agent sunitinib in
metastatic castration-resistant PCa (mCRPC) have suggested that the
antitumor activity, which was assessed by a >50% decline in
prostate-specific antigen levels and tumor shrinkage, exhibited an
acceptable safety profile (18,30). In
addition, several drugs with anti-angiogenic properties have
proceeded to Phase III evaluation for the treatment of patients
with CRPC; however, no phase III study to date has succeeded in
demonstrating a survival benefit in large randomized studies
(54), which indicates that further
advances are required.
In conclusion, the present study demonstrated that
sunitinib manifested an antineoplastic effect on PCa cell lines
in vitro. Sunitinib was revealed to stimulate autophagy in
PCa cells via the regulation of ERK1/2 phosphorylation and mTOR
signaling. Autophagic and apoptotic cell death were demonstrated to
have roles in the cytotoxic effects induced by sunitinib in PCa
cells. Further studies are required to fully investigate the
interactions and signal pathways involved in the conversion between
autophagic and apoptotic cell death. In addition, the findings of
the present study suggest that implementing autophagy combined
sunitinib treatment may be beneficial in devising novel anti-cancer
strategies in PCa.
Acknowledgements
This study was partly supported by the China
National Natural Science Foundation (grant no. 81172421), Ministry
of Science and Technology of China (grant no. 2013CB835300),
Natural Science Foundation of Guangdong Province (grant no.
S2012010010009) and Science and Technology Project of Guangdong
Province (grant no. 2011B031800199).
References
|
1
|
Cuzick J, Thorat MA, Andriole G, Brawley
OW, Brown PH, Culig Z, Eeles RA, Ford LG, Hamdy FC, Holmberg L, et
al: Prevention and early detection of prostate cancer. Lancet
Oncol. 15:e484–e492. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gilligan T and Kantoff PW: Chemotherapy
for prostate cancer. Urology. 60 3 Suppl 1:S94–S100. 2002.
View Article : Google Scholar
|
|
3
|
Thakur MK and Vaishampayan U: Multifaceted
and personalized therapy of advanced prostate cancer. Curr Opin
Oncol. 28:222–231. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lum JJ, Bauer DE, Kong M, Harris MH, Li C,
Lindsten T and Thompson CB: Growth factor regulation of autophagy
and cell survival in the absence of apoptosis. Cell. 120:237–248.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tanida I: Autophagosome formation and
molecular mechanism of autophagy. Antioxid Redox Signal.
14:2201–2214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bursch W: The autophagosomal-lysosomal
compartment in programmed cell death. Cell Death Differ. 8:569–581.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pyo JO, Nah J and Jung YK: Molecules and
their functions in autophagy. Exp Mol Med. 44:73–80. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death-apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Coppin C, Kollmannsberger C, Le L,
Porzsolt F and Wilt TJ: Targeted therapy for advanced renal cell
cancer (RCC): A Cochrane systematic review of published randomised
trials. BJU Int. 108:1556–1563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gori S, Foglietta J, Rossi M, Hamzaj A,
Stocchi L, Galuppo C, Picece V, Puxeddu E and Furlani L: Sunitinib
therapy in metastatic papillary thyroid cancer. Tumori.
99:285e–287e. 2013.PubMed/NCBI
|
|
13
|
O'Reilly EM, Niedzwiecki D, Hall M, Hollis
D, Bekaii-Saab T, Pluard T, Douglas K, Abou-Alfa GK, Kindler HL,
Schilsky RL, et al: A cancer and leukemia group B phase II study of
sunitinib malate in patients with previously treated metastatic
pancreatic adenocarcinoma (CALGB 80603). Oncologist. 15:1310–1319.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reni M, Cereda S, Milella M, Novarino A,
Passardi A, Mambrini A, Di Lucca G, Aprile G, Belli C, Danova M, et
al: Maintenance sunitinib or observation in metastatic pancreatic
adenocarcinoma: A phase II randomised trial. Eur J Cancer.
49:3609–3615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Uno F, Fujiwara Y and Fujiwara T: A
long-term control of gastrointestinal stromal tumor with sunitinib.
Gan To Kagaku Ryoho. 40:1241–1244. 2013.PubMed/NCBI
|
|
16
|
Broxterman HJ, Gotink KJ and Verheul HM:
Understanding the causes of multidrug resistance in cancer: A
comparison of doxorubicin and sunitinib. Drug Resist Updat.
12:114–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fuereder T, Jaeger-Lansky A, Hoeflmayer D,
Preusser M, Strommer S, Cejka D, Koehrer S, Crevenna R and Wacheck
V: mTOR inhibition by everolimus counteracts VEGF induction by
sunitinib and improves anti-tumor activity against gastric cancer
in vivo. Cancer Lett. 296:249–256. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Michaelson M Dror, Regan MM, Oh WK,
Kaufman DS, Olivier K, Michaelson SZ, Spicer B, Gurski C, Kantoff
PW and Smith MR: Phase II study of sunitinib in men with advanced
prostate cancer. Ann Oncol. 20:913–920. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jing C, Ning J and Yuanjie N: The
preventative effects of sunitinib malate observed in the course
from non-castration to castration LNCaP xenograft prostate tumors.
J Cancer Res Clin Oncol. 138:2137–2143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carlisle B, Demko N, Freeman G, Hakala A,
MacKinnon N, Ramsay T, Hey S, London AJ and Kimmelman J: Benefit,
Risk, and outcomes in drug development: A systematic review of
sunitinib. J Natl Cancer Inst. 108:djv2922015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Basch E, Loblaw DA, Oliver TK, Carducci M,
Chen RC, Frame JN, Garrels K, Hotte S, Kattan MW, Raghavan D, et
al: Systemic therapy in men with metastatic castration-resistant
prostate cancer: American society of clinical oncology and cancer
care ontario clinical practice guideline. J Clin Oncol.
32:3436–3448. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deretic V and Levine B: Autophagy,
immunity, and microbial adaptations. Cell Host Microbe. 5:527–549.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen S, Rehman SK, Zhang W, Wen A, Yao L
and Zhang J: Autophagy is a therapeutic target in anticancer drug
resistance. Biochim Biophys Acta. 1806:220–229. 2010.PubMed/NCBI
|
|
24
|
Jin S and White E: Role of autophagy in
cancer: Management of metabolic stress. Autophagy. 3:28–31. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Favata MF, Horiuchi KY, Manos EJ, Daulerio
AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F,
et al: Identification of a novel inhibitor of mitogen-activated
protein kinase kinase. J Biol Chem. 273:18623–18632. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fukazawa H, Noguchi K, Murakami Y and
Uehara Y: Mitogen-activated protein/extracellular signal-regulated
kinase kinase (MEK) inhibitors restore anoikis sensitivity in human
breast cancer cell lines with a constitutively activated
extracellular-regulated kinase (ERK) pathway. Mol Cancer Ther.
1:303–309. 2002.PubMed/NCBI
|
|
27
|
Johnson VL, Ko SC, Holmstrom TH, Eriksson
JE and Chow SC: Effector caspases are dispensable for the early
nuclear morphological changes during chemical-induced apoptosis. J
Cell Sci. 113:2941–2953. 2000.PubMed/NCBI
|
|
28
|
Motzer RJ, Hutson TE, Tomczak P,
Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik
C, Kim ST, et al: Sunitinib versus interferon alfa in metastatic
renal-cell carcinoma. N Engl J Med. 356:115–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Demetri GD, van Oosterom AT, Garrett CR,
Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich
MC, Morgan JA, et al: Efficacy and safety of sunitinib in patients
with advanced gastrointestinal stromal tumour after failure of
imatinib: A randomised controlled trial. Lancet. 368:1329–1338.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sonpavde G, Periman PO, Bernold D,
Weckstein D, Fleming MT, Galsky MD, Berry WR, Zhan F, Boehm KA,
Asmar L and Hutson TE: Sunitinib malate for metastatic
castration-resistant prostate cancer following docetaxel-based
chemotherapy. Ann Oncol. 21:319–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Diaz R, Nguewa PA, Redrado M, Manrique I
and Calvo A: Sunitinib reduces tumor hypoxia and angiogenesis, and
radiosensitizes prostate cancer stem-like cells. Prostate.
75:1137–1149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang ZH, Li Q, Ruan SQ, Xiao Q, Liu Y, Hu
YT, Hu LF, Chen HY, Zheng S, Zhang SZ and Ding KF: Sunitinib
mesylate inhibits proliferation of human colonic stromal
fibroblasts in vitroin vivo. J Zhejiang Univ Sci B. 15:701–712.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Di Desidero T, Fioravanti A, Orlandi P,
Canu B, Giannini R, Borrelli N, Man S, Xu P, Fontanini G, Basolo F,
et al: Antiproliferative and proapoptotic activity of sunitinib on
endothelial and anaplastic thyroid cancer cells via inhibition of
Akt and ERK1/2 phosphorylation and by down-regulation of cyclin-D1.
J Clin Endocrinol Metab. 98:E1465–E1473. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptotic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bolt AM, Zhao F, Pacheco S and Klimecki
WT: Arsenite-induced autophagy is associated with proteotoxicity in
human lymphoblastoid cells. Toxicol Appl Pharmacol. 264:255–261.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Klionsky DJ: Coming soon to a journal near
you-the updated guidelines for the use and interpretation of assays
for monitoring autophagy. Autophagy. 10:16912014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ikeda T, Ishii KA, Saito Y, Miura M,
Otagiri A, Kawakami Y, Shimano H, Hara H and Takekoshi K:
Inhibition of autophagy enhances sunitinib-induced cytotoxicity in
rat pheochromocytoma PC12 cells. J Pharmacol Sci. 121:67–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Santoni M, Amantini C, Morelli MB,
Liberati S, Farfariello V, Nabissi M, Bonfili L, Eleuteri AM,
Mozzicafreddo M, Burattini L, et al: Pazopanib and sunitinib
trigger autophagic and non-autophagic death of bladder tumour
cells. Br J Cancer. 109:1040–1050. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang SH, Shih YL, Ko WC, Wei YH and Shih
CM: Cadmium-induced autophagy and apoptosis are mediated by a
calcium signaling pathway. Cell Mol Life Sci. 65:3640–3652. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sivaprasad U and Basu A: Inhibition of ERK
attenuates autophagy and potentiates tumour necrosis
factor-alpha-induced cell death in MCF-7 cells. J Cell Mol Med.
12:1265–1271. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Corcelle E, Nebout M, Bekri S, Gauthier N,
Hofman P, Poujeol P, Fénichel P and Mograbi B: Disruption of
autophagy at the maturation step by the carcinogen lindane is
associated with the sustained mitogen-activated protein
kinase/extracellular signal-regulated kinase activity. Cancer Res.
66:6861–6870. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fenton MS, Marion KM, Salem AK, Hogen R,
Naeim F and Hershman JM: Sunitinib inhibits MEK/ERK and SAPK/JNK
pathways and increases sodium/iodide symporter expression in
papillary thyroid cancer. Thyroid. 20:965–974. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Voce P, D'Agostino M, Moretti S,
Sponziello M, Rhoden K, Calcinaro F, Tamburrano G, Tallini G,
Puxeddu E, Filetti S, et al: Sunitinib inhibits tumor vascularity
and growth but does not affect Akt and ERK phosphorylation in
xenograft tumors. Oncol Rep. 26:1075–1080. 2011.PubMed/NCBI
|
|
46
|
Lin CI, Whang EE, Moalem J and Ruan DT:
Strategic combination therapy overcomes tyrosine kinase
coactivation in adrenocortical carcinoma. Surgery. 152:1045–1050.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nazio F, Strappazzon F, Antonioli M,
Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J,
Piacentini M, Fimia GM and Cecconi F: mTOR inhibits autophagy by
controlling ULK1 ubiquitylation, self-association and function
through AMBRA1 and TRAF6. Nat Cell Biol. 15:406–416. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hac A, Domachowska A, Narajczyk M, Cyske
K, Pawlik A and Herman-Antosiewicz A: S6K1 controls autophagosome
maturation in autophagy induced by sulforaphane or serum
deprivation. Eur J Cell Biol. 94:470–481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yan L, Vatner DE, Kim SJ, Ge H, Masurekar
M, Massover WH, Yang G, Matsui Y, Sadoshima J and Vatner SF:
Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci
USA. 102:13807–13812. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Notte A, Leclere L and Michiels C:
Autophagy as a mediator of chemotherapy-induced cell death in
cancer. Biochem Pharmacol. 82:427–434. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Donato AL, Huang Q, Liu X, Li F, Zimmerman
MA and Li CY: Caspase 3 promotes surviving melanoma tumor cell
growth after cytotoxic therapy. J Invest Dermatol. 134:1686–1692.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Beltran H, Kaur G, de España CG and Tagawa
ST: Exploring the role of anti-angiogenic therapies in prostate
cancer: Results from the phase 3 trial of sunitinib. Asian J
Androl. 16:568–569. 2014. View Article : Google Scholar : PubMed/NCBI
|