Introduction
MicroRNAs (miRNAs) are an endogenous group of small
(18–25 nucleotides), non-coding RNA molecules which serve key roles
as post-transcriptional regulators by binding to the
3′-untranslated region (3′UTR) of target mRNA (1). Recent results indicate that miRNAs may
modulate numerous cellular processes, including differentiation,
proliferation, apoptosis and migration (2). Thus, miRNAs are considered to be
potential regulators in the development and progression of various
human diseases, including cardiovascular disease, diabetes and
cancer (3–5).
Hepatocellular carcinoma (HCC) is among the most
severe forms of human cancer and is the third-leading cause of
cancer-related mortality worldwide (6). Although treatments for HCC have
improved, the long-term prognosis for patients with HCC remains
poor, with a current 5-year survival rate of ~30% (7,8). A lack
of biomarkers for early diagnosis and effective therapeutic targets
are primary reasons for the poor disease outcomes. Therefore,
studies into the molecular mechanisms of HCC pathogenesis are
warranted, in order to identify potential biomarkers and
therapeutic targets of HCC (9,10).
Recently, the roles of miRNAs in HCC have been investigated, with
results indicating that the expression and/or function of miRNAs
become aberrant in the pathogenesis of HCC (7).
miR-33a is a member of the highly conserved miR-33
family and is an intronic miRNA located within the genes of sterol
regulatory element-binding proteins (11,12),
where it principally regulates the metabolism of cholesterol
(13) and glucose (14). In addition to its functional roles in
metabolism, miR-33a has been implicated in a number of human
cancers. In pancreatic ductal adenocarcinoma, it has been observed
that miR-33a exerts tumor suppressive effects, by modulating the
growth, apoptosis, epithelial-to-mesenchymal transition and
chemoresistance of pancreatic cancer cells (15,16).
Similarly, a previous study in lung cancer demonstrated that
miR-33a had inhibitory effects on the metastasis of cancer cells
towards bone tissue (17). It has
also been observed in glioma cancer that miR-33a promotes the
growth and self-renewal of glioma-initiating cells (18), while previous microarray results have
indicated that miR-33a is upregulated in supraglottic carcinoma
(19). In osteosarcoma (OS), miR-33a
is upregulated in chemoresistant OS and promotes resistance of OS
cells to cisplatin through downregulation of the transcription
factor, Twist (20). Furthermore,
levels of miR-33a and miR-224 expression were elevated in steatotic
chronic hepatitis C when compared to control liver tissue (21), and miR-33a in liver tissue has been
found to significantly increase in a fibrosis progression-dependent
manner (22). Collectively, these
data indicate an oncogenic role of miR-33a, though its clinical
significance and potential roles in HCC remain unknown.
Therefore, the present study investigated the
effects of miR-33a and its underlying mechanisms of action in HCC
using BrdU and apoptosis assays as well as dual luciferase reporter
assays.
Materials and methods
Human tissues and cell culture
A total of 86 individual primary HCC tissues were
collected between January 2010 and January 2012 from patients at
The First Affiliated Hospital of Xi'an Jiaotong University (Xi'an,
China). Patients were monitored for a median time of 31.6 months
(range, 2–60 months). Clinical features of the 86 patients are
listed in Table I. Age, gender, HBV
infection, serum AFP level, tumor size and number of tumor nodules
were measured prior to surgery, while other parameters were
collected after surgery. Clinical specimens were immediately
snap-frozen in liquid nitrogen prior to histological examination.
Exclusion criteria included patients who had received chemotherapy
or embolization prior to surgical resections. Samples were attained
after obtaining informed consent from all patients and all
protocols in the present study were approved by the Medicine Ethics
Committee of Xi'an Jiaotong University in accordance with the
Declaration of Helsinki (as revised in Tokyo 2004) (23).
 | Table I.Clinical association analysis of
miR-33a expression in HCC. |
Table I.
Clinical association analysis of
miR-33a expression in HCC.
|
|
| No. of patients
(n=86) |
|
|---|
|
|
|
|
|
|---|
| Clinicopathological
features | No. of patients
(n=86) | Low miR-33a | High miR-33a | P-value |
|---|
| Age (years) |
|
|
|
|
|
<50 | 27 | 16 | 11 | 0.245 |
|
≥50 | 59 | 27 | 32 |
|
| Gender |
|
|
|
|
|
Male | 69 | 34 | 35 | 0.787 |
|
Female | 17 | 9 | 8 |
|
| HBV |
|
|
|
|
|
Absent | 30 | 14 | 16 | 0.651 |
|
Present | 56 | 29 | 27 |
|
| Serum AFP level,
ng/ml |
|
|
|
|
|
<20 | 20 | 11 | 9 | 0.610 |
|
≥20 | 66 | 32 | 34 |
|
| Tumor size, cm |
|
|
|
|
|
<5 | 30 | 20 | 10 | 0.024 |
| ≥5 | 56 | 23 | 33 |
|
| No. of tumor
nodules |
|
|
|
|
| 1 | 66 | 35 | 31 | 0.307 |
| ≥2 | 20 | 8 | 12 |
|
| Cirrhosis |
|
|
|
|
|
Absent | 37 | 22 | 15 | 0.127 |
|
Present | 49 | 21 | 28 |
|
| Venous
infiltration |
|
|
|
|
|
Absent | 42 | 23 | 19 | 0.388 |
|
Present | 44 | 20 | 24 |
|
| Edmondson-Steiner
grading |
|
|
|
|
|
I+II | 49 | 31 | 18 | 0.005 |
|
III+IV | 37 | 12 | 25 |
|
| TNM tumor
stage |
|
|
|
|
|
I+II | 61 | 35 | 26 | 0.033 |
|
III+IV | 25 | 8 | 17 |
|
Human HCC cell lines, HepG2 and Huh7 (Shanghai
Institute of Biochemistry and Cell Biology, Chinese Academy of
Sciences, Shanghai, China), were cultured in complete Dulbecco's
modified Eagle's medium (Biosera, Inc., Villebon sur Yvette,
France) supplemented with 10% fetal bovine serum (Biosera, Inc.),
100 units/ml penicillin and 100 µg/ml streptomycin (Sigma-Aldrich,
Merck KGaA, Darmstadt, Germany). Cell lines were cultured in a
humidified atmosphere in a 5% CO2 incubator at 37°C for
2–3 days and collected for further analyses.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from frozen tissues or
cultured HCC cells using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
instructions. Quantification of miR-33a was performed using a
TaqMan MicroRNA Assay kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and U6 small nuclear RNA was used as an
endogenous control by determining the fold-change in miR-33a
expression relative to U6 expression. For PPARα quantification,
cDNA was synthesized using Taqman RT reagents (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Total RNA (2 µg) was reverse
transcribed at 37°C for 15 min, and cDNA was incubated at 85°C for
5 sec to inactivate the reverse transcriptase. cDNA (2 µl) was used
for the qPCR, which was performed using a SYBR Premix Ex Taq II
Perfect Real Time kit (Takara Bio, Inc., Otsu, Japan) in the ABI
PRISM 7300 Sequence Detection system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The reactions were incubated at 95°C for
60 sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 34
sec. Expression of PPARα mRNA was normalized to that of GAPDH and
the primer sequences were as follows: For PPARα forward,
5′-ACTGTTGCAAGAGATCTACAGAG-3′ and reverse,
5′-TTGTCTGTCACTGTCTGAATCC-3′ and for GAPDH forward,
5′-AACTTTGGCATTGTGGAAGG-3′ and reverse, 5′-ACACATTGGGGGTAGGAACA-3′.
All samples were normalized to internal controls and fold changes
were calculated based on relative quantification using the
2−ΔΔCq method (24).
Cell transfection
Overexpression and inhibition of miR-33a in Huh7 and
HepG2 cells, respectively, was established using miRNA vectors
obtained from GeneCopoeia, Inc. (Rockville, MD, USA). In accordance
with the manufacturer's instructions for Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), Huh7 cells at 75%
confluence were transfected with an miR-33a expression vector
(HmiR0366-MR03) or an miRNA scrambled control vector
(CmiR0001-MR03), and HepG2 cells were transfected with an miR-33a
inhibitory vector (HmiR-AN0429-AM03B) or a negative non-coding
vector (CmiR-AN0001-SN). The efficacy of miR-33a or anti-miR-33a
vector transfection was assessed by RT-qPCR as outlined above. The
primer sequences used for miR-33a were as follows: forward,
5′-CGCGCGTGCATTGTAGTTG-3′ and reverse, 5′-CACCAGGGTCCGAGGT-3′ and
stem loop primer,
5′-TGGATATCCACACCAGGGTCCGAGGTATTCGGTGTGGATATCCATGCAATG-3′.
Cell proliferation and apoptosis
assays
For the proliferation assay, transfected HCC cells
were seeded into 96-well plates (1×103 cells/well) and
incubated at 37°C. At 48 h post-transfection, the proliferative
ability of HCC cells was assessed using a 5-bromo-2-deoxyuridine
(BrdU) Cell Proliferation ELISA kit (Roche Diagnostics,
Indianapolis, IN, USA), according to the manufacturer's
instructions. For the apoptosis assay, HCC cells were seeded into
6-well plates (6×104 cells/well) and incubated at 37°C.
At 48 h post-transfection, an Annexin-V-FLUOS Staining kit (Roche
Diagnostics) was used to determine the percentage of apoptotic
cells by flow cytometry, according to the manufacturer's
instructions. Flow cytometric analysis was conducted using
fluorescence-activated cell sorting Calibur (BD Biosciences, San
Jose, CA, USA) and Cell Quest Pro v.4.0.2 software (BD
Biosciences). Similar results were obtained in three independent
experiments performed in duplicate.
Dual luciferase reporter assay
A dual luciferase reporter assay was performed to
determine whether PPARα was a downstream target gene of miR-33a in
HCC cells. Briefly, the 3′-UTR sequence of PPARα, which was
predicted to interact with miR-33a using two publicly available
databases (TargetScan 6.2, targetscan.org; miRanDa, microrna.org),
or a mutated sequence within the predicted sites were synthesized
(GeneChem Co., Ltd., Shanghai, China) and inserted into the XbaI
and FseI restriction sites of a pGL3 control vector (luciferase
reporter vector; Promega Corporation, Madison, WI, USA) downstream
of a luciferase minigene, as previously reported (25). These constructs were named as
wild-type (wt) PPARα-3′UTR or mutant (mt) PPARα-3′UTR,
respectively. For the reporter assay, HepG2 cells that were seeded
into 96-well plates (5×103 cells/well) were cultured in
complete Dulbecco's modified Eagle medium (Biosera, Inc.)
supplemented with 10% fetal bovine serum (Biosera, Inc.) at 37°C
and co-transfected with miRNA mimics or inhibitors as outlined
above, the above constructs and Renilla plasmid (Promega
Corporation) using FuGENE HD Transfection Reagent (Promega
Corporation), according to the manufacturer's protocol. At 48 h
post-transfection, cells were harvested and Renilla and firefly
luciferase activities were quantified using a Dual Luciferase Assay
system (Promega Corporation), according to the manufacturer's
protocol. Firefly luciferase activity was normalized to that of
Renilla luciferase. Results were obtained from three independent
experiments performed in triplicate.
Immunoblotting
HCC cells were lysed in radioimmunoprecipitation
assay buffer (50 mM Tris pH 7.5, 150 mM sodium chloride, 1% Triton
X-100, 5 mM ethylenediaminetetraacetic acid) at 4°C for 1 h, then
insoluble material was removed by centrifugation at 12,000 × g for
10 min. A total of 30 µg of the resulting protein samples (per
lane) were separated by 4–12% SDS-PAGE and transferred onto a
nitrocellulose membrane. Blots were then incubated with primary
antibodies against PPARα (sc-398394; 1:1,000; Santa Cruz
Technology, Inc., Santa Cruz, CA, USA) and GAPDH (5174; 1:1,500;
Cell Signaling Technology, Inc., Danvers, MA, USA) at 4°C for at
least 12 h. After three washes with Tris-buffered saline-Tween-20,
blots were incubated with horseradish peroxidase-conjugated goat
anti-mouse (sc-2005; 1:5,000; Santa Cruz Technology, Inc.) or
anti-rabbit secondary antibodies (1662408; 1:10,000; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) at room temperature for 2 h,
detected using a Bio-Rad Gel imaging system and quantified using
Quantity One v.4.1 software (both from Bio-Rad Laboratories,
Inc.).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Group comparisons were conducted using a two-sample
t-test or one-way analysis of variance (multiple comparisons).
Categorical variables were compared using χ2 analysis or
Fisher's exact test. Kaplan-Meier analysis was used to analyze
overall survival and recurrence-free survival. SPSS 13.0 (SPSS,
Inc., Chicago, IL, USA) and GraphPad Prism 5.0 software (GraphPad
Software, Inc., La Jolla, CA, USA) were used for statistical
analysis and P<0.05 (two-tailed) was considered to indicate a
statistically significant difference.
Results
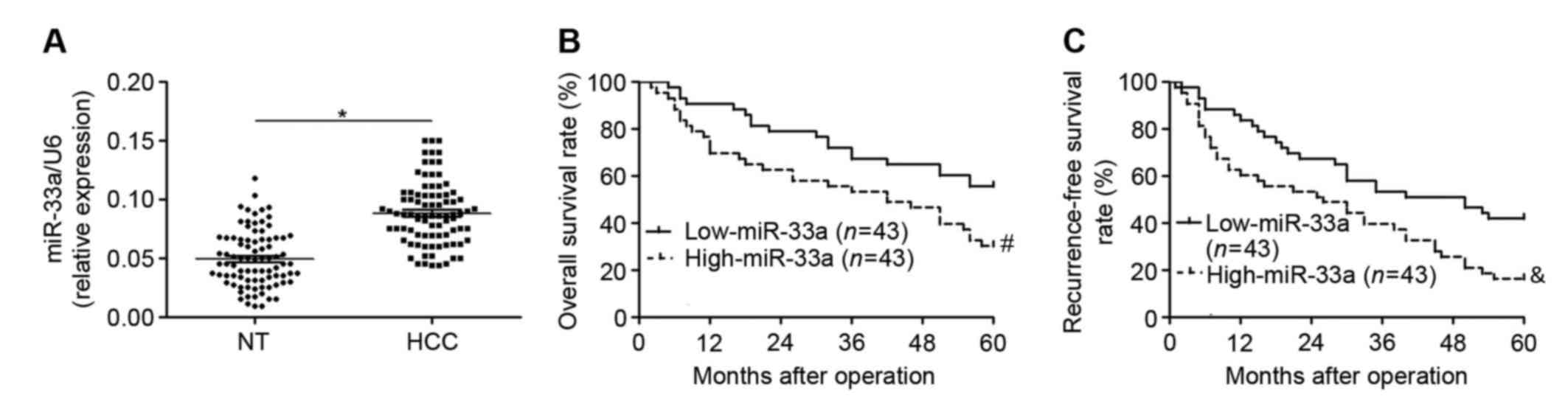
Increased miR-33a expression in HCC
correlates with adverse clinical features and poor prognosis
To elucidate the expression status and clinical
significance of miR-33a in HCC, the levels of miR-33a were measured
in HCC tissues and matched adjacent non-tumor tissues from 86 HCC
patients. It was observed that levels of miR-33a were significantly
higher in HCC tissues relative to adjacent non-tumor tissues
(P<0.05; Fig. 1A). The clinical
significance of miR-33a expression in HCC patients was subsequently
investigated. Expression of miR-33a in HCC patients was determined
to be low (n=43) or high (n=43) according to a cutoff value, which
was defined as the median level of miR-33a in the patient cohort
(0.087). As depicted in Table I,
high levels of miR-33a expression were significantly correlated
with larger tumor size (P=0.024), higher Edmondson-Steiner grading
(poor differentiation; P=0.005) and higher tumor-node-metastasis
tumor stage (P=0.033). Furthermore, a Kaplan-Meier analysis
demonstrated that patients with high levels of miR-33a exhibited
significant decreases in overall survival rate (P=0.015; Fig. 1B) and recurrence-free survival rate
(P=0.008; Fig. 1C). These results
suggest that miR-33a serves an oncogenic role and may be a
prognostic indicator in HCC.
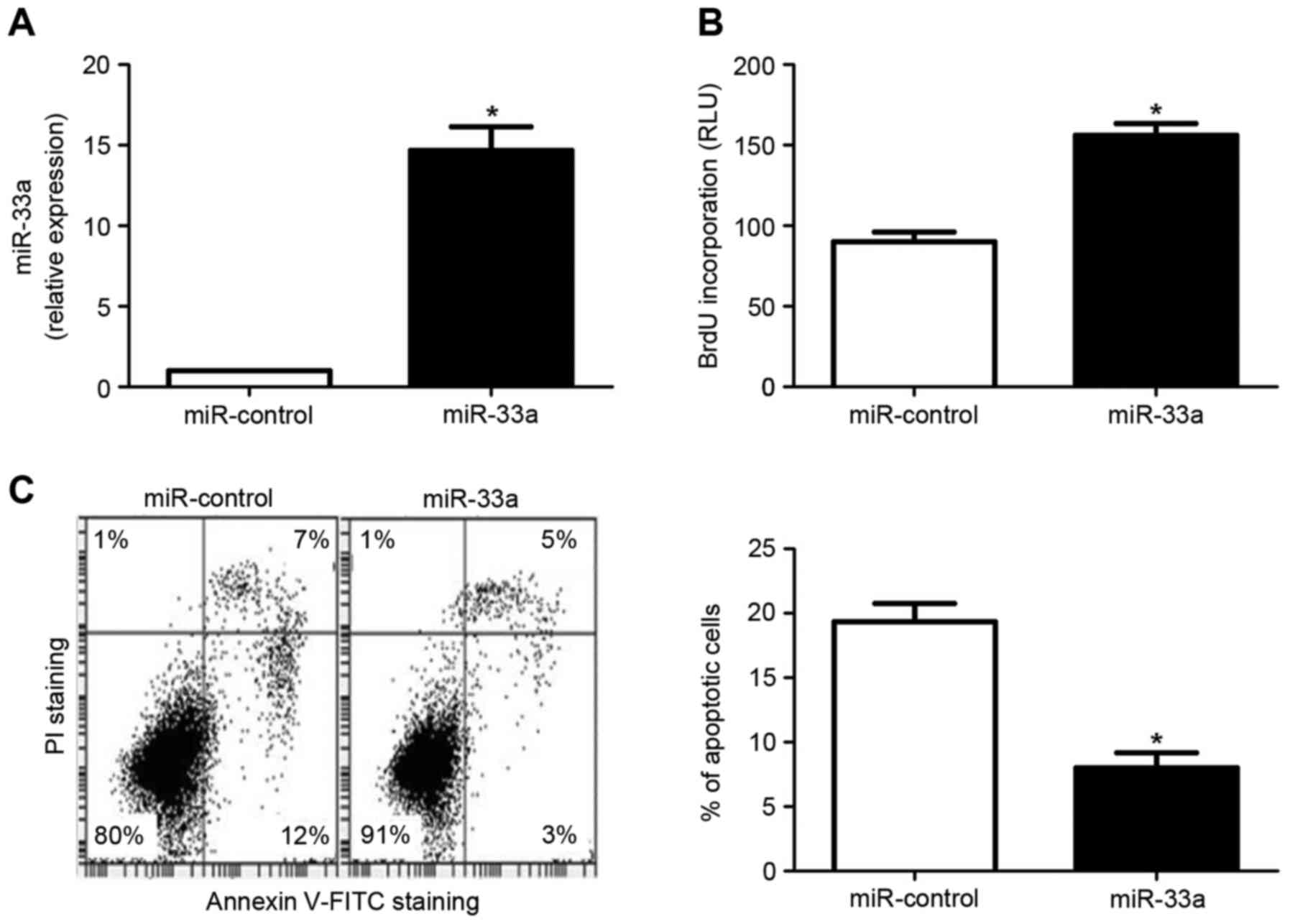
miR-33a induces proliferation and
inhibits apoptosis in HCC cells
As increased proliferation and reduced apoptosis are
key hallmarks of cancer cells (26),
the oncogenic effects of miR-133a were subsequently determined by
evaluating its effects on the proliferation and apoptosis of HCC
cells. The HCC cell line Huh7 was transfected with an miR-33a
expression vector or an miR control vector. As depicted in Fig. 2A, miR-33a expression was
significantly upregulated in Huh7 cells transfected with the
miR-33a expression vector, relative to cells transfected with the
miR control vector (P<0.05). Subsequently, a BrdU incorporation
assay indicated that forced expression of miR-33a in Huh7 cells led
to a significant increase in cellular proliferation (P<0.05;
Fig. 2B). In addition, an Annexin
V/propidium iodide double staining assay demonstrated that the rate
of apoptosis was significantly decreased in Huh7 cells
overexpressing miR-33a (P<0.05; Fig.
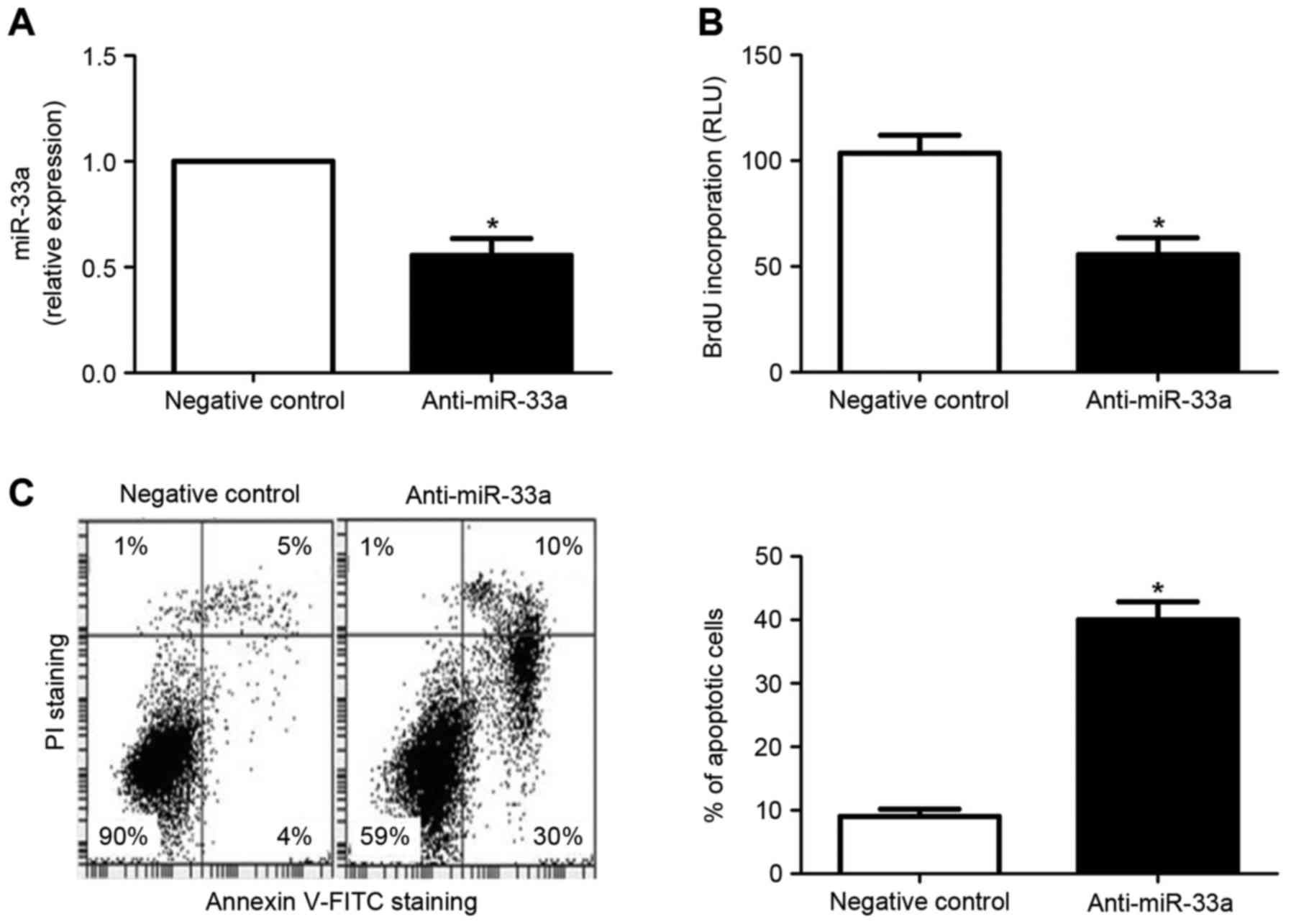
2C). By contrast, transfection of the HCC cell line HepG2 with
miR-33a inhibitors resulted in significantly decreased miR-33a
expression, relative to cells transfected with a negative control
vector (P<0.05; Fig. 3A). In
turn, miR-33a downregulation lead to significantly decreased
proliferation (P<0.05; Fig. 3B)
and increased apoptosis (P<0.05; Fig.
3C) in HepG2 cells. Collectively, these results indicate
miR-33a may promote the development and progression of HCC by
potentiating proliferation and inhibiting apoptosis of HCC
cells.
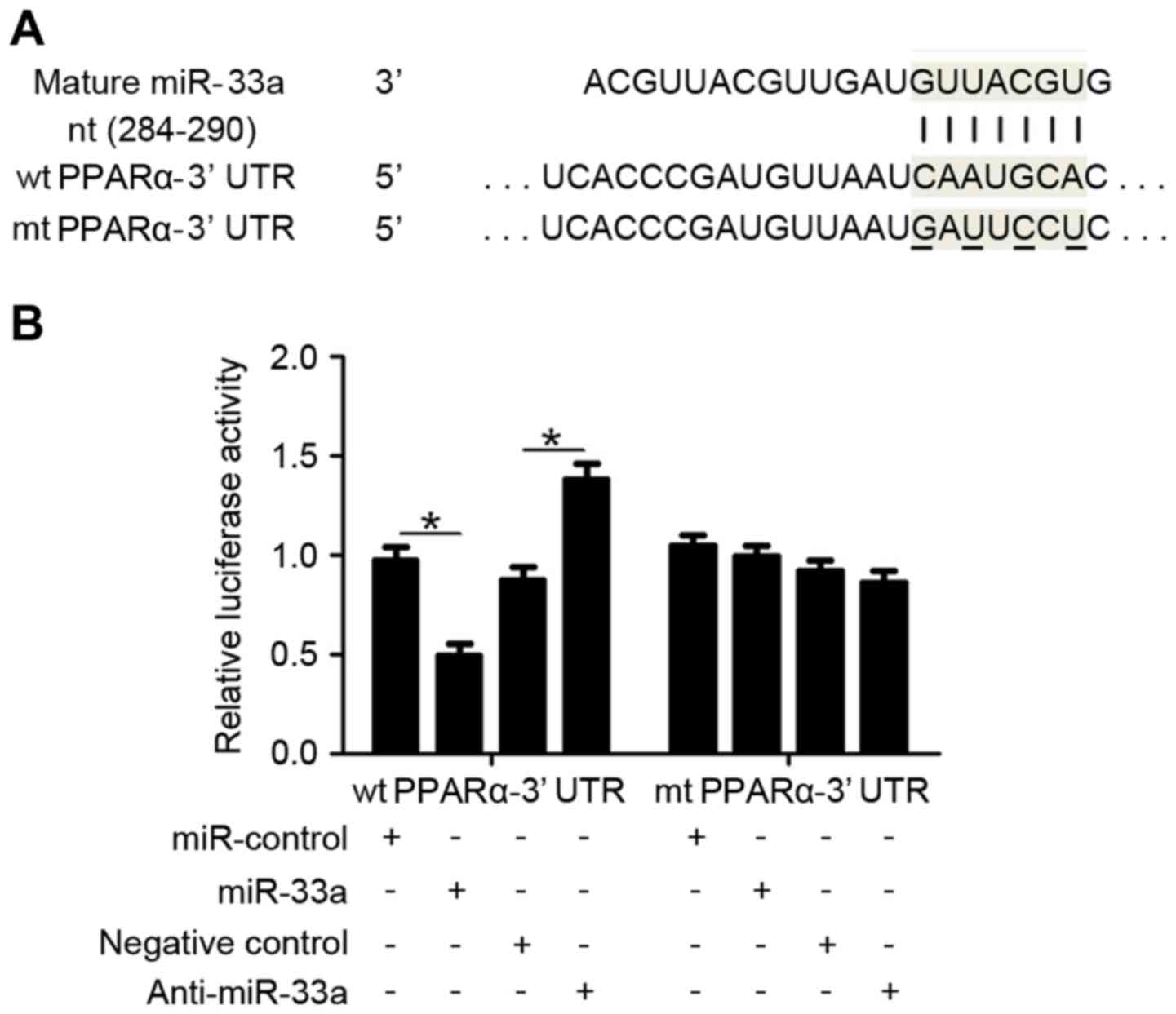
PPARα is a direct downstream target of
miR-33a in HCC cells
To identify the underlying mechanisms by which
miR-33a exerts its potential oncogenic effects in HCC cells, two
publicly available databases, TargetScan 6.2 (www.targetscan.org) and miRanDa (www.microrna.org), were used to predict the target
sequences of miR-33a. PPARα, which is considered to be a key
regulator of HCC cell proliferation and apoptosis (27), was identified as a miR-33a target. As
depicted in Fig. 4A, the 3′-UTR of
PPARα mRNA contains a complementary sequence for miR-33a binding,
suggesting that PPARα is a direct downstream target of miR-33a.
Dual-luciferase reporter gene assays were subsequently performed to
confirm whether miR-33a targets the 3′-UTR of PPARα mRNA, using
wild type (wt) and mutant (mt) PPARα-3′UTRs (Fig. 4A). As shown in Fig. 4B, overexpression of miR-33a in HepG2
cells significantly inhibited the luciferase activity of PPARα
expressing a wt 3′-UTR (P<0.05), while having no effect on that
of mt PPARα-3′UTR. Accordingly, downregulation of miR-33a in HepG2
cells lead to significantly increased luciferase activity of wt
PPARα-3′UTR (P<0.05), while having no significant effect on that
of mt PPARα-3′UTR. These results indicate that PPARα is a direct
downstream target of miR-33a.
miR-33a regulates the expression of
PPARα in HCC cells
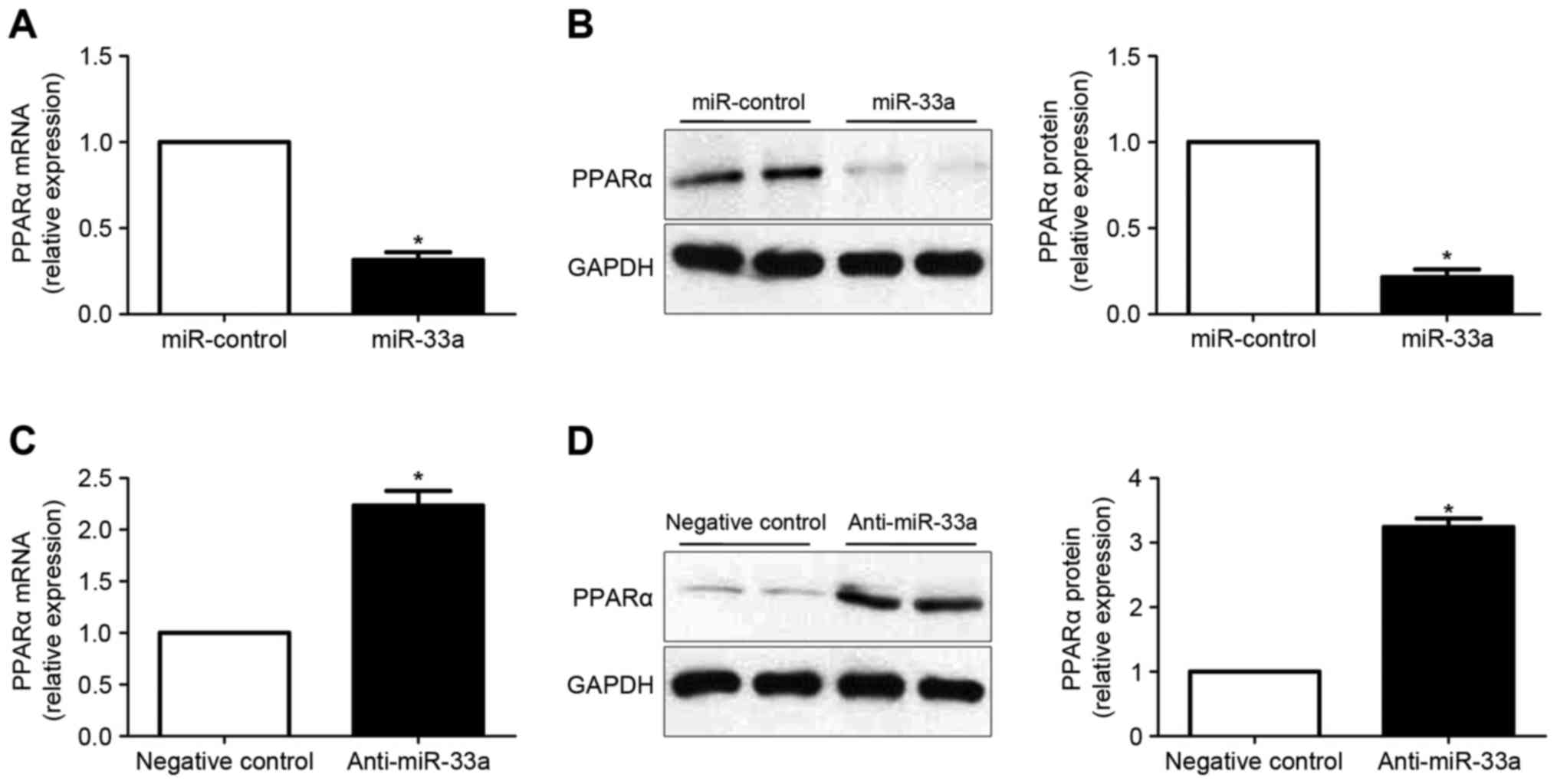
RT-qPCR and western blot analysis were subsequently
performed to determine whether miR-33a regulates the expression of
PPARα in HCC cells. It was observed that overexpression of miR-33a
in Huh7 cells significantly decreased the levels of PPARα mRNA
(P<0.05; Fig. 5A). Results of
western blot analysis also demonstrated that the levels of PPARα
protein was significantly reduced following forced expression of
miR-33a (P<0.05; Fig. 5B). By
contrast, downregulation of miR-33a in HepG2 cells lead to
significantly increased PPARα expression at the mRNA (P<0.05;
Fig. 5C) and protein (P<0.05,
Fig. 5D) levels.
Discussion
miRNAs regulate gene expression at the
post-transcriptional level and have been demonstrated to
participate in numerous biological processes, including
embryogenesis, differentiation, morphogenesis and tumorigenesis
(1,3,4,28). In the last two decades, studies have
investigated the potential roles of miRNAs in cancer. It has been
indicated that miRNAs may have oncogenic or tumor-suppressive
effects in human malignancies and may also serve as promising
biomarkers and therapeutic targets in the diagnosis and treatment
of cancer (3,29). miR-33a is an established regulator of
glucose and cholesterol metabolism (14,30) and
has been demonstrated to be an active regulator in the pathogenesis
of various human cancers, including pancreatic cancer (15,16),
lung cancer (17), glioma (18), osteosarcoma (31) and colon cancer (32). In the present study, significant
overexpression of miR-33a was confirmed in HCC tissues. In turn,
increased expression of miR-33a was associated with adverse
clinical features and a poor prognosis for HCC patients. These
results indicate that miR-33a may serve an oncogenic role in HCC
and as a potential biomarker for the diagnosis and prognostic
prediction of HCC.
Functionally, miR-33a is a key regulator of
cholesterol metabolism through its manipulation of adenosine
triphosphate-binding cassette transporter A1 levels (13). In addition, it regulates glucose
metabolism through targeting of phosphoenolpyruvate carboxykinase
and glucose-6-phosphatase (14).
However, miR-33a is also implicated in numerous aspects of cancer
biology. In pancreatic cancer cells, miR-33a has been demonstrated
to enhance gemcitabine sensitivity (16), while in lung cancer, the miRNA acts a
bone metastasis suppressor through its targeting of parathyroid
hormone related protein (17). It
has also been suggested that miR-33a may promote the self-renewal
of glioma-initiating cells (18). In
the present study, gain- and loss-of-function experiments
demonstrated that miR-33a may promote the growth of HCC by
potentiating proliferation and inhibiting apoptosis of HCC cells.
Specifically, overexpression of miR-33a promoted proliferation and
inhibited apoptosis of Huh7 cells, while inhibition of miR-33a
decreased proliferation and increased apoptosis of HepG2 cells.
Therefore, these results indicate that miR-33a may promote the
development and progression of HCC, at least in part through
modulation of cell proliferation and apoptosis.
PPARα is a member of the nuclear hormone receptor
superfamily and participates in the metabolism of glucose and
lipids (33). In addition, it has
been identified as a tumor suppressor in colorectal carcinoma
(34) and ovarian cancer (35). Regarding HCC, a previous study has
demonstrated that PPARα exerted anti-tumorigenic effects in HCC
cells through modulation of nuclear factor-κB signaling (27). It has also been observed in HCC cells
that ectopic expression of PPARα significantly suppressed
proliferation while inducing apoptosis (27). In the present study, a complementary
sequence for miR-33a was identified in the 3′-UTR of PPARα. In
turn, alterations in miR-33a expression led to significant changes
in luciferase activity of wt PPARα-3′-UTR, while having no
influence on that of mt PPARα-3′-UTR. Furthermore, ectopic
expression of miR-33a significantly reduced the expression of
PPARα, while downregulation of miR-33a led to a significant
increase in PPARα expression. Collectively, these data indicate
that PPARα is a direct downstream target of miR-33a in HCC cells.
Thus, the regulatory effects of miR-33a on HCC cell proliferation
and apoptosis may be due to its targeting of PPARα.
In conclusion, the present study demonstrated that
miR-33a is overexpressed in HCC tissues, with elevated expression
of miR-33a correlated with adverse clinical features and poor
prognosis in HCC. Functional experiments also demonstrated that
miR-33a may promote cell growth by modulating the proliferation and
apoptosis of HCC cells. Furthermore, PPARα was verified to be a
direct downstream target of miR-33a. Collectively, these data
suggest that miR-33a may be a novel clinical biomarker for the
diagnosis and prognosis of HCC, as well as a potential therapeutic
target for the treatment of HCC.
References
|
1
|
Yates LA, Norbury CJ and Gilbert RJ: The
long and short of microRNA. Cell. 153:516–519. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Osman A: MicroRNAs in health and
disease-basic science and clinical applications. Clin Lab.
58:393–402. 2012.PubMed/NCBI
|
|
5
|
Rottiers V and Näär AM: MicroRNAs in
metabolism and metabolic disorders. Nat Rev Mol Cell Biol.
13:239–250. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang N, Ekanem NR, Sakyi CA and Ray SD:
Hepatocellular carcinoma and microRNA: New perspectives on
therapeutics and diagnostics. Adv Drug Deliv Rev. 81:62–74. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dhanasekaran R, Limaye A and Cabrera R:
Hepatocellular carcinoma: Current trends in worldwide epidemiology,
risk factors, diagnosis, and therapeutics. Hepat Med. 4:19–37.
2012.PubMed/NCBI
|
|
9
|
Aravalli RN, Steer CJ and Cressman EN:
Molecular mechanisms of hepatocellular carcinoma. Hepatology.
48:2047–2063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tanaka S and Arii S: Molecular targeted
therapies in hepatocellular carcinoma. Semin Oncol. 39:486–492.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cirera-Salinas D, Pauta M, Allen RM,
Salerno AG, Ramírez CM, Chamorro-Jorganes A, Wanschel AC, Lasuncion
MA, Morales-Ruiz M, Suarez Y, et al: Mir-33 regulates cell
proliferation and cell cycle progression. Cell Cycle. 11:922–933.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li T, Francl JM, Boehme S and Chiang JY:
Regulation of cholesterol and bile acid homeostasis by the
cholesterol 7α-hydroxylase/steroid response element-binding protein
2/microRNA-33a axis in mice. Hepatology. 58:1111–1121. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Najafi-Shoushtari SH, Kristo F, Li Y,
Shioda T, Cohen DE, Gerszten RE and Näär AM: MicroRNA-33 and the
SREBP host genes cooperate to control cholesterol homeostasis.
Science. 328:1566–1569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ramírez CM, Goedeke L, Rotllan N, Yoon JH,
Cirera-Salinas D, Mattison JA, Suárez Y, de Cabo R, Gorospe M and
Fernández-Hernando C: MicroRNA 33 regulates glucose metabolism. Mol
Cell Biol. 33:2891–2902. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang C, Yu XJ, Guo XZ, Sun MH, Wang Z,
Song Y, Ni QX, Li HY, Mukaida N and Li YY: MicroRNA-33a-mediated
downregulation of Pim-3 kinase expression renders human pancreatic
cancer cells sensitivity to gemcitabine. Oncotarget. 6:14440–14455.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang C, Wang Z, Li YY, Yu BH, Zhang F and
Li HY: miR-33a suppresses the nuclear translocation of β-catenin to
enhance gemcitabine sensitivity in human pancreatic cancer cells.
Tumour Biol. 36:9395–9403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kuo PL, Liao SH, Hung JY, Huang MS and Hsu
YL: MicroRNA-33a functions as a bone metastasis suppressor in lung
cancer by targeting parathyroid hormone related protein. Biochim
Biophys Acta. 1830:3756–3766. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang H, Sun T, Hu J, Zhang R, Rao Y, Wang
S, Chen R, McLendon RE, Friedman AH, Keir ST, et al: miR-33a
promotes glioma-initiating cell self-renewal via PKA and NOTCH
pathways. J Clin Invest. 124:4489–4502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang T, Han G, Wang Y, Chen K and Sun Y:
MicroRNA expression profiles in supraglottic carcinoma. Oncol Rep.
31:2029–2034. 2014.PubMed/NCBI
|
|
20
|
Zhou Y, Huang Z, Wu S, Zang X, Liu M and
Shi J: miR-33a is up-regulated in chemoresistant osteosarcoma and
promotes osteosarcoma cell resistance to cisplatin by
down-regulating TWIST. J Exp Clin Cancer Res. 33:122014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lendvai G, Jármay K, Karácsony G, Halász
T, Kovalszky I, Baghy K, Wittmann T, Schaff Z and Kiss A: Elevated
miR-33a and miR-224 in steatotic chronic hepatitis C liver
biopsies. World J Gastroenterol. 20:15343–15350. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang CF, Sun CC, Zhao F, Zhang YD and Li
DJ: miR-33a levels in hepatic and serum after chronic HBV-induced
fibrosis. J Gastroenterol. 50:480–490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
World Medical Association, . World Medical
Association Declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C (T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shimono Y, Zabala M, Cho RW, Lobo N,
Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, et al:
Downregulation of miRNA-200c links breast cancer stem cells with
normal stem cells. Cell. 138:592–603. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang N, Chu ES, Zhang J, Li X, Liang Q,
Chen J, Chen M, Teoh N, Farrell G, Sung JJ and Yu J: Peroxisome
proliferator activated receptor alpha inhibits hepatocarcinogenesis
through mediating NF-κB signaling pathway. Oncotarget. 5:8330–8340.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lujambio A and Lowe SW: The microcosmos of
cancer. Nature. 482:347–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cho WC: MicroRNAs: Potential biomarkers
for cancer diagnosis, prognosis and targets for therapy. Int J
Biochem Cell Biol. 42:1273–1281. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dávalos A, Goedeke L, Smibert P, Ramírez
CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U,
Pastor-Pareja JC, et al: miR-33a/b contribute to the regulation of
fatty acid metabolism and insulin signaling. Proc Natl Acad Sci
USA. 108:9232–9237. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou Y, Huang ZF, Wu S, Zang XF, Liu M and
Shi J: miR-33a is up-regulated in chemoresistant osteosarcoma and
promotes osteosarcoma cell resistance to cisplatin by
down-regulating TWIST. J Exp Clin Canc Res. 33:122014. View Article : Google Scholar
|
|
32
|
Ibrahim AF, Weirauch U, Thomas M,
Grünweller A, Hartmann RK and Aigner A: MicroRNA replacement
therapy for miR-145 and miR-33a is efficacious in a model of colon
carcinoma. Cancer Res. 71:5214–5224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lefebvre P, Chinetti G, Fruchart JC and
Staels B: Sorting out the roles of PPAR alpha in energy metabolism
and vascular homeostasis. J Clin Invest. 116:571–580. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grau R, Punzón C, Fresno M and Iñiguez MA:
Peroxisome-proliferator-activated receptor alpha agonists inhibit
cyclo-oxygenase 2 and vascular endothelial growth factor
transcriptional activation in human colorectal carcinoma cells via
inhibition of activator protein-1. Biochem J. 395:81–88. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yokoyama Y, Xin B, Shigeto T, Umemoto M,
Kasai-Sakamoto A, Futagami M, Tsuchida S, Al-Mulla F and Mizunuma
H: Clofibric acid, a peroxisome proliferator-activated receptor
alpha ligand, inhibits growth of human ovarian cancer. Mol Cancer
Ther. 6:1379–1386. 2007. View Article : Google Scholar : PubMed/NCBI
|