Introduction
Previous laboratory and clinical studies suggest
that reperfusion following myocardial infarction may induce
ischemia/reperfusion injury (1–15).
Lethal reperfusion injuries are hypothesized to account for up to
half of the final infarction area (2). Myocardial ischemia/reperfusion injury
is a frequent clinical problem (13–19).
Despite optimal myocardial reperfusion, the mortality rate after
acute myocardial infarction approaches 10% (19). This is the main pathophysiological
basis for cardiac insufficiency and arrhythmia following angina and
a number of clinical treatments (such as early reperfusion therapy
of acute myocardial infarction and cardiac transplantation), and is
the focus of much current research. Ischemic preconditioning and
postconditioning have been therapeutically validated in various
ischemia/reperfusion injury animal models, and several clinical
trials (16–18). Currently, ischemic events cannot be
predicted in clinical practice; hence, ischemic and pharmacological
preconditioning is less implementable. However, ischemic
postconditioning has its own pitfalls; repetitive inflations and
deflations of the heart during percutaneous coronary angioplasty
may lead to coronary endothelial damages, plaque rupture or
dislodgement, coronary artery rupture and intervention
complications (20). Pharmacological
postconditioning via a simulative endogenous protective mechanism
(following ischemia and prior to reperfusion) is more practicable
due to its effectiveness, safety and easy manipulation (8,21).
Statins, inhibitors of reductase and
hydroxymethylglutaryl-CoA (HMG-CoA) reductase, are frequently used
to lower blood lipids, especially cholesterol, and have proven
cardioprotective effects (22–26).
Besides their lipid-lowering properties, statins have been
demonstrated to exert extrahepatic, cholesterol-independent
effects, or ‘pleiotropic’ effects, in previous animal studies
(22,23,25,27),
including improved endothelial function, altered inflammatory
responses, maintenance of plaque stability and prevention of
thrombus formation. In animal models of ischemia/reperfusion
injury, all members of the statin family including atorvastatin
calcium, have been proven to reduce the myocardial infarction size
(22,23,25,28,29).
Common cardioprotective strategies such as ischemic
preconditioning (IPC) and ischemic postconditioning (Ipost) have
also been reported to limit the size of myocardial infarction in
young, healthy male animals (8,10,30–35).
However, additional experiments have revealed that the protection
may be impaired or removed in animals dependent on aging,
hyperglycemic state, hypertension or hypercholesterolemia. The
mortality of diabetic animals and patients following reperfusion
was reported to be many times higher than that of non-diabetic
animals or patients undergoing the same treatment; this may be
partly caused by the inhibition of IPC- and Ipost-mediated
protective mechanisms that occurs in diabetes (7,12,36,37).
It was previously reported that IPC and Ipost mediate myocardial
protection by stimulating phosphoinositide 3-kinase (PI3K)/Akt and
the associated glycogen synthase kinase (GSK3β) pathway. Increasing
evidence indicates that PI3K/Akt and the associated GSK-3β pathway
is inhibited in diabetes, implying that diabetes attenuated IPC-and
Ipost-mediated myocardial protection may occur via these pathways
(36,38–40).
Theoretically, any strategy to reactivate PI3K/Akt and the
associated GSK-3β pathway may exert a protective effect against
ischemia/reperfusion injuries in diabetes.
Therefore, the present study was designed to address
the hypothesis that atorvastatin calcium may provide a protective
effect against reperfusion injury in STZ-induced diabetes by
phosphorylating GSK-3β.
Materials and methods
Animals and materials
A total of 96 male Sprague-Dawley (SD) rats
(Shanghai SLAC Laboratory Animal Co., Ltd., Shanghai, China), 3–4
weeks of age, and weighing 50±5 g, were used in this study (n=12
per group) which conformed to the Guide for the Care and Use of
Laboratory Animals published by the US National Institutes of
Health guidelines regulating the care and use of laboratory animals
and was approved by the Experimental Animal Care Committee of
Fujian Medical University Union Hospital (Fuzhou, China).
Atorvastatin calcium was obtained from Dailan Melone Biotechnology
Co., Ltd. (Dalian, China). Primary antibodies against phospho-GSK3β
(p-GSK3β; ab131097), total GSK3β (ab124661), and heat shock factor
(HSF)-1 (ab131081) were purchased from Abcam (Cambridge, UK).
Secondary antibodies (ZB-2301) and primary antibodies against GAPDH
(YT5052) were purchased from Beijing Chinese Fir Golden Bridge
Biotechnology Co., Ltd. (Beijing, China). Serum total cholesterol
(TC; 467852), triglyceride (TG; 445850), heat shock protein (HSP)70
(CSB-E08308r) and cardiac troponin I (cTnI; S795) were determined
using commercially available ELISA kits (Stanbio Laboratory,
Boerne, TX, USA), in accordance with the manufacturer's
instructions. Evan's blue, triphenyltetrazolium chloride (TTC),
dimethyl sulfoxide and streptozotocin, were purchased from
Sigma-Aldrich (Merck KGaA; Darmstadt, Germany).
Induction of diabetes
Previous studies have reported different
susceptibilities of the hearts of patients with diabetes to
reperfusion injury compared with patients without diabetes
(24,39). Prior to the development of a
myocardial ischemia/reperfusion injury model, vulnerability to
myocardial ischemia/reperfusion injury in the streptozotocin
(STZ)-induced diabetic rat model was examined. The experimental SD
rats were housed in standard polypropylene cages (4 rats/cage)
under a 12/12-h light/dark cycle and an ambient temperature of
22–25°C. Animals were randomly divided into two groups, and fed
either a regular chow, as a control, or a high-fat diet. Regular
chow consisted of 60% carbohydrate, 12% fat, and 28% protein and
the high-fat diet consisted of 41% carbohydrate, 40% fat, and 18%
protein. After 4 weeks, 1% STZ (45 mg/kg, dissolved in 0.1 mmol/l
citrate buffer pH 4.5–4.6) was injected into the abdominal cavity
of high-fat diet group rats to create the SD rat model of diabetic
mellitus, and the rats from the control group were injected with
0.1 mmol/l citrate buffer (4.5 ml/kg). STZ-injected animals were
provided with access to food and water ad libitum following
the STZ injection, and both STZ-injected and non-injected animals
continued their individual diets. At 72 h after injection, the
blood glucose levels were tested using a glucose meter (Optium
Xceed; Abbott Pharmaceutical Co. Ltd., Lake Bluff, IL, USA) and
rats with blood glucose levels>11 mmol/l were determined to be
diabetic.
Measurements of general
characteristics
Water intake and food consumption were evaluated
daily, body weight was monitored weekly. The blood glucose levels
were tested once a week using a glucose meter. At termination (4
weeks after injection), rats were weighed subsequent to an
overnight fast of 8–10 h, and were anesthetized with 75 mg/kg
ketamine and 7.5 mg/kg diazepam intraperitoneally. Blood samples
were obtained from the abdominal aorta after 180 min reperfusion,
and the separated plasma was stored at −80°C until assayed. Serum
TC, TG, HSP70 and cTnI were determined using the aforementioned
kits.
Animal grouping and treatments
Diabetic rats (DM) were randomly divided into six
groups (n=12/group), and age-matched male non-diabetic SD rats
(NDM) were randomly divided into two groups (n=12/group).
Non-diabetic rats were randomly assigned to the sham or
ischemia/reperfusion (I/R) groups. Diabetic rats were divided into
groups as follows: i) Sham; ii) I/R; iii) TDZD-8 +optimal dose (2
mg/kg) of atorvastatin calcium (T+AP); iv) AP1, atorvastatin
calcium postconditioning (1 mg/kg); v) AP2, atorvastatin calcium
postconditioning (2 mg/kg); and vi) DMSO groups. The sham operation
(Sham) group was treated with open chest operation but without
myocardial ischemia/reperfusion. The study design is outlined in
Fig. 1.
Myocardial ischemia reperfusion injury
in vivo
The experimental rats' body temperature was
maintained at 37°C during surgery, which was performed under
anesthesia. Rats were placed in a supine position and mechanical
ventilation was maintained with a rodent respirator (Jiangxi Teli
Anaesthesia & Respiration Equipment Co., Ltd., Jiangxi, China)
with a tidal volume of 1.0 ml/100 mg body weight (60 breaths/min)
in accordance with a previous study (10). An electrocardiogram (ECG) monitor was
connected to provide long-term, continuous and real-time ECG
information. A left thoracotomy was performed on the fourth
intercostal space, and the thoracic cavity was exposed by blunt
dissection. The pericardial tissue was removed, a single 5–0
Prolene suture was placed under the LCA, 1–2 mm from its origin. A
small polyethylene tube was placed between the ends for reversible
coronary artery occlusion. Rats received 40 min of LAD occlusion
followed by 180 min of reperfusion. The sham group rats were
treated in the same manner, with the exception that the suture was
not ligated.
Blood collection and tissue
harvest
Following reperfusion, blood samples were obtained
immediately. The heart was removed and the myocardium was divided
into two parts: One was fixed with 10% formalin, paraffin-embedded,
made into slices for hematoxylin and eosin staining or
immunohistochemical analysis; the other was stored at −70°C for
western-blot analysis.
Determination of area at risk and
infarct size
LAD was reoccluded in situ at the end of
reperfusion, 1.5–2 ml of 3% Evan's blue dye was injected into the
right ventricle via the venous system. Cardiac arrest in diastole
was induced by an intravenous bolus injection of 10% KCl solution.
The heart tissue was kept at −20°C until it was in a semi-frozen
state such that it was easily sliced. The frozen heart was
transversely cut into slices with a thickness of 2 mm. The slices
were then incubated in a 1% solution of TTC and agitated at a
temperature of 37°C for 10 to 15 min. Following staining and
fixation (in 10% formalin for 24 h), these were arranged
sequentially, imaged by digital camera and quantified using ImageJ
v. 1.36 (imagej.nih.gov/ij/). The area of
necrosis (AN; uncolored), area at risk (AAR; including the
uncolored and red area) and the left ventricular area (LV) were
measured. The infarct size was expressed by AN/AAR and the risk
area was expressed by AAR/LV.
Histochemical analysis
Pathological characterization of the heart was
performed by histology and immunohistochemical staining. After
reperfusion, the heart was removed, and the ventricular wall was
removed from the site of ligation (about 3×5 mm in size) for HE
staining. For immunohistochemical staining, the myocardial
specimens were harvested after 5 min reperfusion, and
deparaffinized tissue sections were treated with 0.3% hydrogen
peroxide to block endogenous peroxidase activity. Tissue sections
were then incubated in a protein-free blocking agent (Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA) for 10 min to inhibit the
nonspecific binding of primary antibodies. Sections were examined
for the presence and distribution of p-GSK3β. The dilution ratios
of the primary antibodies (anti-GSK3β, anti-p-GSK3β and anti-HSF-1)
and the goat anti-rabbit secondary antibody were 1:900, 1:700,
1:1,200 and 1:1,000, respectively. Incubationw ith the primary
antibodies was performed for 2 h at 37°C and the secondary antibody
was incubated for 30 min at 37°C.
Immunostains were examined using a microscope
(Leica, Munich, Germany), and digital images were analyzed using
Image-Pro Plus software (version 6; Media Cybernetics Inc.,
Bethesda, MA, USA).
cTnI and HSP70 levels
Expression levels of these proteins were measured
using rat ELISA kits in accordance with the manufacturer's
instructions.
Western blot analysis
After 15 min reperfusion, hearts were quickly
obtained for subsequent protein immunoblot analysis. The samples
(60 µg protein/lane) were separated using 12% sodium dodecyl
sulfate polyacrylamide gel electrophoresis, and then transferred
from the gel onto Hybond-C Extra membranes (Amersham, Pittsburgh,
PA, USA). To prevent the non-specific binding, the membrane was
placed in a membrane block, such as 3–5% bovine serum albumin.
Following blocking of non-specific binding, the membranes were
sequentially incubated with primary antibodies (anti- GSK3β,
anti-p-GSK3β and anti-HSF-1) and the secondary antibody
(Sigma-Aldrich). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
served as the internal reference. The immunoreactive bands were
detected by Enhanced Chemiluminescence Plus reagent (GE Healthcare
Life Sciences, Chalfont, UK) using an X-ray film (Kodak, Rochester,
NY, USA). Target signals were assessed using ImageJ 1.36
software.
Statistical analysis
Data were analyzed using SPSS software (version
13.0; SPSS Inc., Chicago, IL, USA). All values were continuous
variables with normal distribution and were expressed as the mean ±
standard deviation (SD). One-way analysis of variance was used for
statistical analyses of data obtained within the same group of rats
and between groups of rats, and this was followed by Tukey's test
for multiple comparisons of group means. A P-value of <0.05 was
considered to indicate a statistically significant difference.
Results
General characteristics at
termination
As presented in Table
I, at 4 weeks after STZ injection, STZ-induced diabetic rats
presented significantly increased water intake, food consumption,
plasma glucose, triglycerides and cholesterol, compared with
control group (all P<0.05). Furthermore, the body weight in the
diabetic group was lower than non-diabetic group (P<0.05).
 | Table I.Baseline characteristics prior to
ischemia/reperfusion. |
Table I.
Baseline characteristics prior to
ischemia/reperfusion.
| Parameters | NDM | DM |
|---|
| Water intake,
ml/kg/day | 120.7±8.6 |
308.3±11.3a |
| Food consumption,
g/kg/day | 60.9±3.8 |
137.6±10.2a |
| Body weight, g | 412.8±13.7 |
355.4±22.9a |
| Plasma glucose,
mM | 5.1±0.8 |
13.8±3.6a |
| Triglycerides,
mg/dl | 120.1±10.4 |
420.4±20.9a |
| Cholesterol,
mg/dl | 78.16±7.7 |
184.6±24.3a |
Evans blue-TTC dyeing
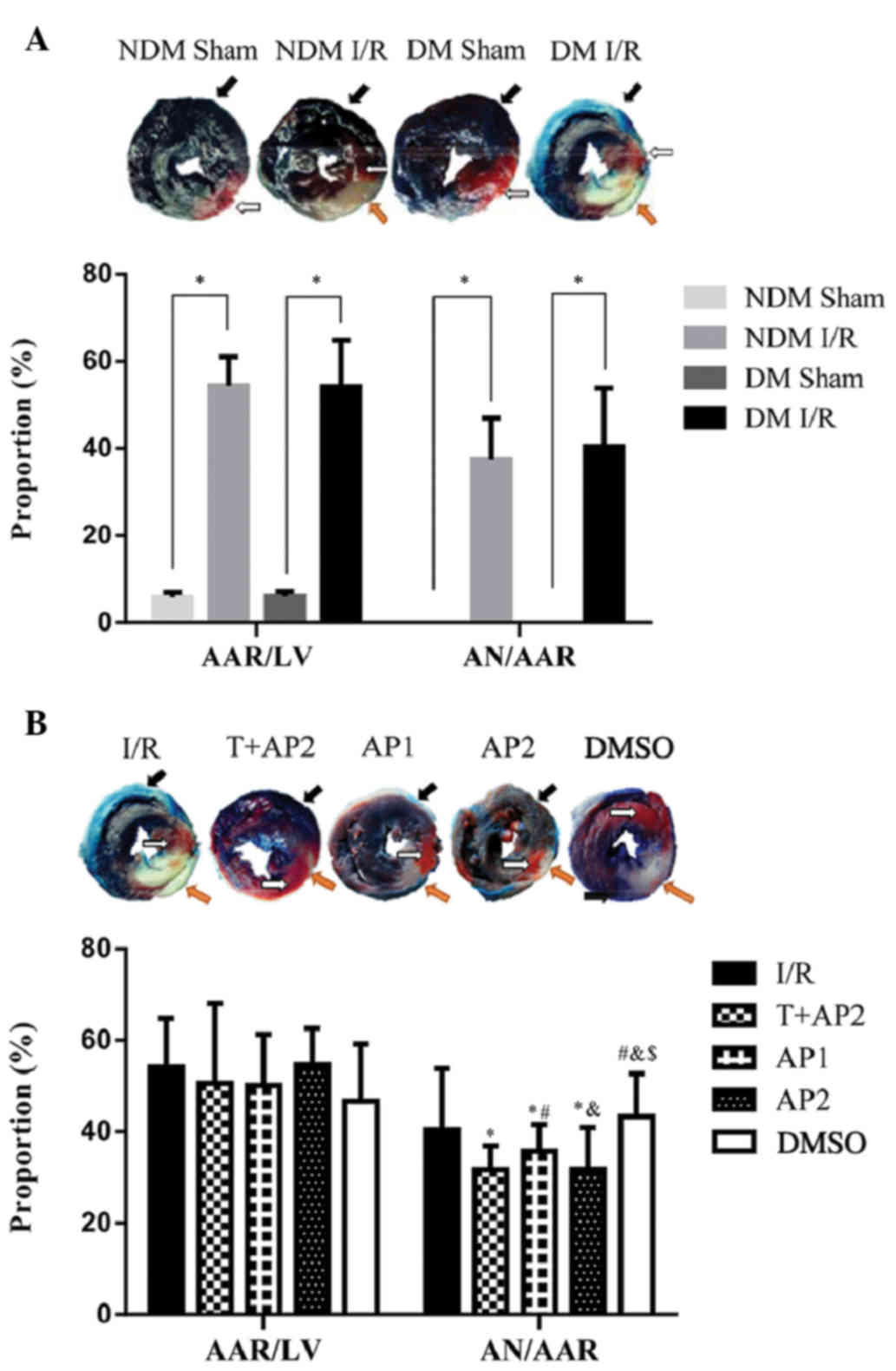
As presented in Fig.
2, the black arrow pointed to the non-ischemic (various tones
of blue), white arrow to AAR (various tones of red), and orange
arrow to necrotic area (pale, uncolored). AAR is expressed as
AAR/LV and the AN is expressed as AN/AAR. Myocardial infarct size
was significantly increased in DM groups subjected to ischemia and
postischemic reperfusion than that in the NDM groups (Fig. 2A). No necrotic areas were observed in
the sham group. No significant difference was identified among the
other groups in AAR/LV (P>0.05). Infarct sizes (AN/AAR) in the
T+AP2, AP1 and AP2 groups were significantly decreased compared
with the the I/R control group (31.71±5.20, 35.68±5.87 and
31.79±9.13 vs. 40.46±13.36%, respectively; P<0.05; Fig. 2B). As shown in Fig. 2B, compared with AP1 group, the AP2
treatment could further reduce myocardial infarct size (35.68±5.87
vs.31.79±9.13%, P<0.05). No significant differences were
observed between the I/R and DMSO groups (40.46±13.36 vs.
43.27±9.40%, P>0.05), nor between the T+AP2 and AP2 groups
(31.71±5.20 vs. 31.79±9.13%; P>0.05). Compared with the I/R
group, the T+AP, AP1 and AP2 groups presented with significantly
reduced infarct sizes (AN/AAR; all P<0.05). The reduction in
infarct size induced by optima dose atorvastatin calcium
postconditioning was not enhanced by TDZD-8 (T+AP2 group), a
specific inhibitor of GSK3β.
Hematoxylin-eosin staining
To evaluate the severity of the cardiomyocyte
injuries in the different groups, both in NDM and DM, morphologic
changes were observed using hematoxylin-eosin staining (Fig. 3A). In the NDM sham group, myocardial
fibers were regularly arranged in order, and no swelling,
denaturation, necrosis, neutrophil infiltration, or other obvious
pathological changes were observed. By contrast, inflammatory cells
were observed in the DM Sham cytoplasm. Other groups, including NDM
I/R and DM I/R, DM T+AP2, DM AP1, DM AP2 and DM DMSO, displayed
varying degrees of morphological lesion. The order of the damage
degree among the six groups, from most serious to least, was as
follows: DM I/R, DMSO, NDM I/R, AP1, while histological changes in
myocytes were least noticable in the T+AP2 and AP2 groups (Fig. 3A).
Immunohistochemical staining of
p-GSK3β
Immunohistochemical staining was used for the
qualitative analysis and the spatial distribution of p-GSK3β.
p-GSK3β positive expression present as brown coloration distributed
in the cytoplasm and nucleus. p-GSK3β was not expressed or weakly
expressed in the DM Sham and DM I/R groups. p-GSK3β was positively
expressed in the NDM, T+AP2, AP1 and AP2 groups, in the cytoplasm
and nucleus. The orders of staining intensities from strong to weak
were as follows: T+AP, AP2, NDM and AP1. However, the expression
levels of p-GSK3β in these groups were not marked (Fig. 3B). p-GSK3β quantitative analysis were
attained by western blot analysis, as described below.
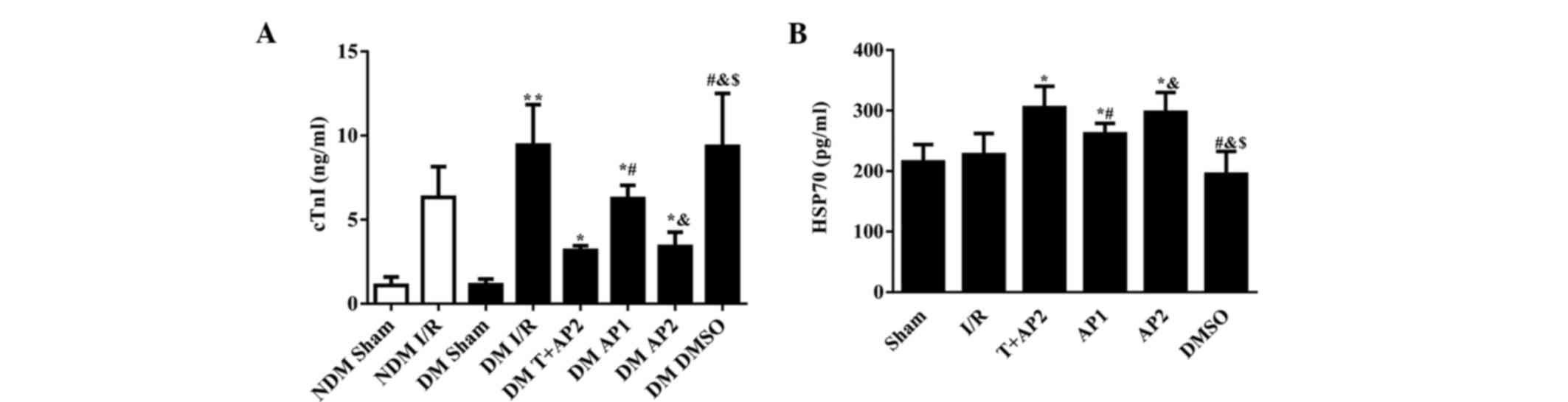
Cardiac troponin I levels in the NDM
and DM groups
As displayed in Fig.
4A, significant differences were detected between the NDM sham
and DM sham groups compared with the corresponding groups suffered
from ischemia reperfusion injury (P<0.01). Following 40 min of
LAD with 180 min reperfusion, the concentration of serum cTnI was
significantly increased in the DM I/R relative to the NDM I/R group
(9.43±2.41 vs. 6.33±1.83, P<0.05). By contrast, no significant
differences were observed between the NDM sham and DM sham groups
(1.09±0.50 vs. 1.13±0.34; P>0.05). DM I/R and DM DMSO groups
(8.74±3.02 vs. 9.36±3.15, P>0.05), nor between the DM T+AP2 and
DM AP2 groups (3.17±0.27 vs. 3.40±0.86, P>0.05). The serum cTnI
concentrations in the DM T+AP2, DM AP1 and DM AP2 groups were
significantly lower than compared with the DM I/R group (3.17±0.27,
6.25±0.79 and 3.40±0.86 vs. 8.74±3.02, respectively; P<0.05).
Compared with the AP1 group, the AP2 group presented with
significantly decreased serum cTnI concentration (3.40±0.86 vs.
6.25±0.79, P<0.05; Fig. 4A).
Serum HSP70 levels in DM groups
As show in Fig. 4B,
the differences in serum HSP70 concentration among all the DM
groups appeared less marked than those in cTnI (Fig. 4A); however, a number of the
inter-group differences reached statistical significance. No
significant differences were observed between the I/R and DMSO
groups (215.01±28.92 vs. 194.93±37.72, P>0.05), nor between the
T+AP2 and AP2 groups (304.92±35.24 vs. 297.04±32.67, P>0.05).
However, the serum HSP70 concentrationsin the T+AP2, AP1 and AP2
groups were significantly higher than in the I/R group
(304.92±35.24, 261.51±17.41 and 297.04±32.67 vs. 227.17±35.08,
respectively; P<0.05). Compared the AP1 group, the AP2 group
displayed significantly increased serum HSP70 levels (297.04±32.67
vs. 261.51±17.41, P<0.05; Fig.
4B).
Western blot analysis
Expression levels of p-GSK3β/t-GSK3β and HSF-1 in
the myocardium were evaluated using western blot analysis (Fig. 5). No statistically significant
difference was observed in HSF-1 levels between the NDM sham and DM
sham groups (Fig. 5C), while the NDM
sham had significantly higher p-GSK3β/t-GSK3β expression ratios
compared with the DM sham (0.37±0.025 vs. 0.20±0.020; P<0.05,
Fig. 5A). Compared with the DM I/R
groups, the NDM I/R group had higher p-GSK3β/t-GSK3β and HSF-1
expression ratios (p-GSK3β/t-GSK3β, 0.45±0.03 vs 0.23±0.024; HSF-1,
2.8±0.30 vs. 1.7±0.31, respectively; all P<0.05, Fig.5A and C).
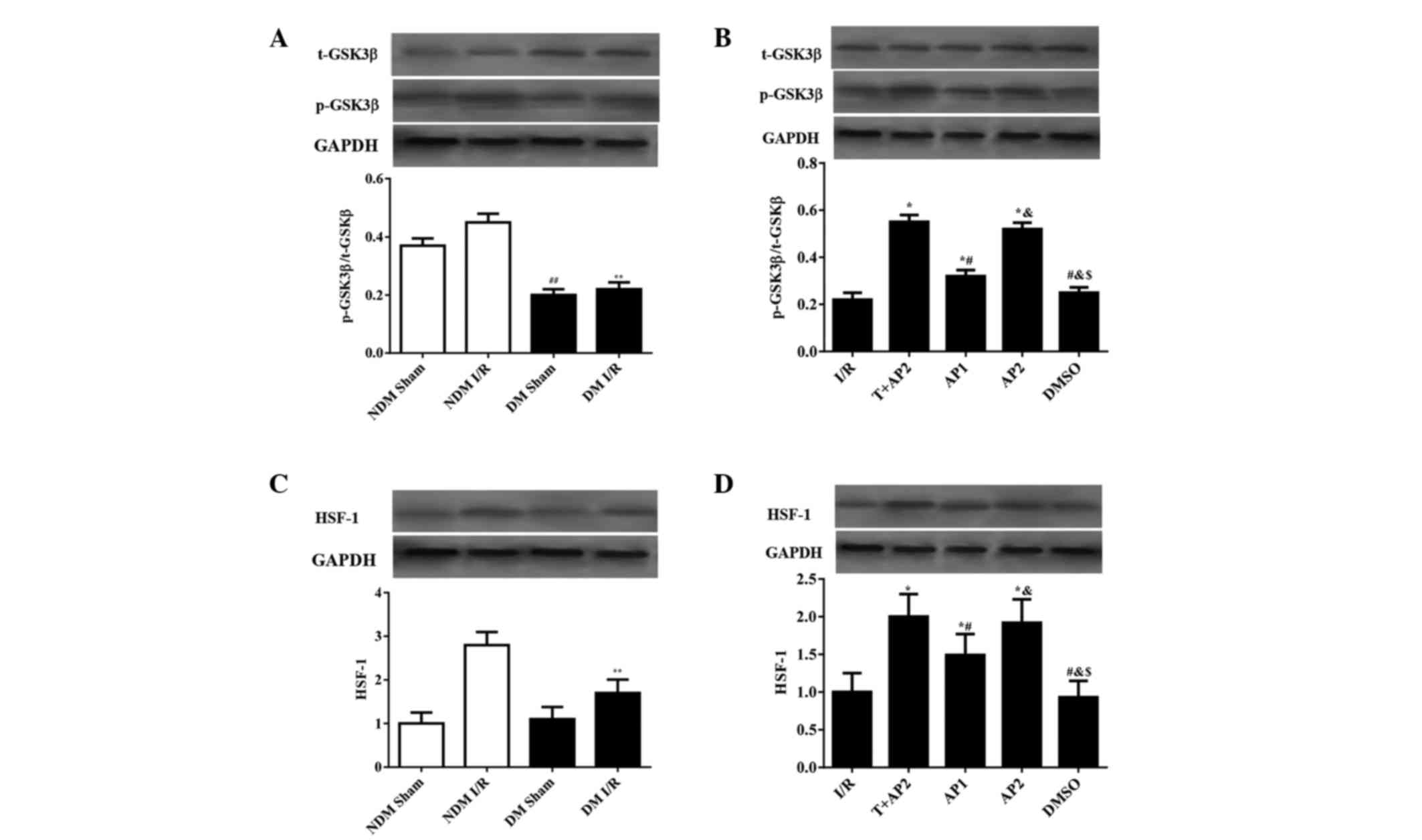 | Figure 5.Western blotting analyses to indicate
(A and B) t-GSK3β and p-GSK3β; and (C and D) HSF-1 in NDM and DM
groups, or DM groups only, respectively. ##P<0.05 vs.
NDM Sham; **P<0.05 vs. NDM I/R; *P<0.05 vs. I/R;
#P<0.05 vs. T + AP2; &P<0.05 vs.
AP1; $P<0.05 vs. AP2. t-GSK3β, total glycogen
synthase kinase 3β; p-GSK3β, phospho-glycogen synthase kinase 3β;
HSF-1, heat shock factor-1; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; NDM, non-diabetic rats; DM, diabetic rats; T,
TDZD-8; AP1, atorvastatin calcium postconditioning (1 mg/kg); AV2,
atorvastatin calcium postconditioning (2 mg/kg). |
As shown in Fig. 5B and
D, the differences in HSF-1 expression among all the DM groups
show similar patterns to the p-GSK3β/t-GSK3β expression ratios. No
significant differences were observed between the I/R and DMSO
groups (p-GSK3β/t-GSK3β, 0.22±0.03 vs. 0.25±0.022; HSF-1, 1.0±0.25
vs. 0.93±0.22, respectively; all P>0.05), nor between the T+AP2
and AP2 groups (p-GSK3β/t-GSK3β, 0.55±0.03 vs 0.52±0.027; HSF-1,
2.0±0.30 vs. 1.92±0.31, respectively; all P>0.05; Fig. 5B and D). The p-GSK3β/t-GSK3β
expression ratios and HSF-1 levels in the T+AP2, AP1 and AP2 groups
were higher compared with the I/R group (p-GSK3β/t-GSK3β,
0.55±0.03, 032±0.026 and 0.52±0.027 vs. 0.22±0.03; HSF-1,
1.92±0.31, 1.49±0.28 and 2.0±0.30 vs. 1.0±0.25, respectively; all
P<0.05). In addition, compared with the AP1 group, the AP2 group
presented with significantly increased p-GSK3β/t-GSK3β expression
ratios and HSF-1 levels (p-GSK3β/t-GSK3β, 032±0.026 vs 0.52±0.027;
HSF-1, 1.0±0.25 vs. 2.0±0.30, respectively; all P<0.05; Fig. 5B and D).
Discussion
Despite the ongoing development of therapies, the
mortality rate of patients with ischemic heart disease remains
high, particularly in cases of DM (7,36,37).
Diabetes is an independent risk factor for myocardial ischemia, and
numerous epidemiological data and laboratory studies have also
revealed that diabetes significantly exacerbated myocardial
ischemia/reperfusion injury and weakened the regular protective
effects (9,12,36,37). The
present study intended to develop a rat model of type 2 diabetes
similar to that in clinical practice. However, in previous studies,
the infarct size in animal models of DM has been reported to be
both different to and similar in size to those of nondiabetic
controls (7,9,11,12,30,41).
Due to differences in experimental design, a single factor would be
insufficient to explain the vast variance in the effects of
diabetes on the infarct size. In the current study, the fasting
blood glucose and the serum TG and TC of diabetic rats were
increased compared with the non-diabetic rats, while the body
weight in these rats did not fall significantly in the first three
weeks after injection of STZ. Following 40 min ischemia and 180 min
reperfusion, no significant difference of the myocardial infarction
size was observed between the non-diabetic rats and diabetic rats,
while in diabetic rats, the levels of serum cTnI, morphological
lesions of myocardial cells, and the level of GSK3β were higher
than in non-diabetic rats. This indicated that the diabetic heart
may be more susceptible to ischemia/reperfusion injury.
Atorvastatin calcium, as the inhibitor of HMG-CoA
reductase, is fat-soluble and is commonly used in the treatment of
cardiovascular diseases. Its cardioprotective effects were
confirmed in numerous previous studies (22,28,29). It
was hypothesized in the current study that atorvastatin calcium
postconditioning alleviates myocardial ischemia/reperfusion injury
on STZ-induced diabetic rats by phosphorylating GSK3β.
In the present study, compared with DM I/R group,
the myocardial infarction size and the levels of cTnI decreased,
and the size of the morphological lesions were attenuated in the
atorvastatin calcium postconditioning groups. Atorvastatin calcium
postconditioning (1 or 2 mg/kg) therefore alleviated myocardial
ischemia/reperfusion injury in STZ-induced diabetic rats. Compared
with the 1 mg/kg atorvastatin calcium postconditioning group, the 2
mg/kg atorvastatin calcium postconditioning group further
alleviated morphological lesions, reduced myocardial infarct size,
reduced the level of cTnI and increased the level of p-GSK3β. It
was therefore demonstrated that 2 mg/kg atorvastatin calcium was
the optimal dose. No significant differences were reported in the
myocardial infarction size, cTnI levels, morphological lesions of
the myocardial cells or the level of p-GSK3β between the
TDZD-8+optimal dose of atorvastatin calcium group and the 2 mg/kg
atorvastatin calcium group. These data suggest no synergy between
TDZD-8 and atorvastatin calcium, and that they may protect the
myocardial cells by the same pathway. A previous study of TDZD-8,
as the specific inhibitor of GSK3β, has demonstrated that GSK-3β
inhibitors protect against myocardial ischemia/reperfusion injury
via inhibition of inflammation and apoptosis (34). Therefore, GSK3β phosphorylation may
serve a vital role during atorvastatin calcium postconditioning.
The changes to HSF-1 and the serum HSP70 levels tend to follow a
similar expression pattern as p-GSK3β in cardiomyocytes, and
p-GSK3β was localized to the plasma and nuclei of cardiomyocytes,
as determined by immunohistochemistical analyses. Concordantly,
p-GSK3β accelerated HSP70 production partially by activating HSF-1
during myocardial ischemia.
GSK3 proteins are serine/threonine kinases, first
identified in 1980 (42), is highly
conserved in evolution. In mammals, GSK-3 proteins include GSK-3α
(51 kDa) and GSK3β (47 kDa), encoded by separate genes, and the
kinase domain has up to 98% homology (39,43).
Nearly 20 years after the discovery, researchers previously
considered that the role of GSK3 was as its name indicates; a
glycogen kinase involved in the phosphorylation of protein kinase
regulation of glucose metabolism. However, an increasing number of
studies have suggested that protein substrates of phosphorylated
GSK3 included ~50 types, including as many as a dozen transcription
factors (31,34,38–40,43–45).
GSK3 serves an important function in the regulation of numerous
cellular functions and activities, such as embryogenesis, cell
proliferation, differentiation, apoptosis, signal transduction and
microtubules movements. Furthermore, GSK3 is involved in the
occurrence and development of a number of diseases, such as
Alzheimer's disease, bipolar disorder, schizophrenia, cancer and
diabetes (39).
GSK-3β is a constitutively active 47-a Ser/Thr
protein kinase, which decreases glycogen synthase activity.
However, GSK-3β is now known as a multifunctional kinase, serving
functions in glycogen metabolism in addition to cell proliferation,
growth and death (39,40,43). In
the cardiovascular system, GSK-3β serves major roles in glucose
metabolism (45), cardiomyocyte
hypertrophy (46) and cell death.
S9-phosphorylation of GSK3β is required for postconditioning, and
is hypothesized to function by inhibiting the opening of the
mitochondrial permeability transition pore (mPTP) (31). Prior studies have indicated that mPTP
is involved in the development of myocardial reperfusion injury.
mPTP, which was first reported by Haworth and Hunter in 1979
(47), is a non-specific channel
located in the mitochondrial membrane, and allows small molecules
(<1.5 kDa) to pass though. mPTP is closed under normal
physiological conditions in order to maintain the integrity of the
structure and function of mitochondria, and remains closed
following ischemia. However, as a result of reperfusion injury,
oxygen free radical levels are increased and calcium overload
activates the opening of mPTPs (32). A prior study indicated that mPTPs
opened at 5–10 min after reperfusion (48), allowing water and solutes into the
mitochondria non-selectively, resulting in mitochondrial membrane
potential imbalance, uncoupling of oxidative phosphorylation,
mitochondrial swelling and ultimately leading to cell death. It is
now generally believed that mPTP is a key factor in myocardial
reperfusion injury, and it could be an important target for
alleviating reperfusion injury (2,21,31,32,38,44,47–49).
GSK3β serves a crucial function in the initial phase
of mPTP opening, which was first reported by Juhaszova et al
in 2004 (44). They found that
inactivating cardiac myocyte GSK3β could increase the mPTP opening
threshold caused by ROS in vitro. Gomez et al
(31) provided further evidence that
the mPTP opening caused by calcium overload in vitro was
inhibited after ischemic postconditioning in wild-type mice, while
in mutated mice whose GSK3β coudl not be phosphorylated and
inactivated, ischemic postconditioning could not inhibit mPTP
opening. Miki et al (38)
suggested that the proportion of phosphorylated GSK3β relative to
total GSK3β in mitochondria is associated with with the threshold
of mPTP opening triggered by calcium ions. Theoretically, GSK3β
inactivation inhibits ROS generation and mPTP opening induced by
calcium overload, and thus reduces myocardial reperfusion injury.
In basic research, the application of GSK3β pharmacological
inhibitor (SB216763 and SB415286) before ischemia or after
reperfusion has been shown to reduce infarct size, and furthermore
ischemic preconditioning, ischemic postconditioning
andcardioprotective drug application all led to the phosphorylation
of GSK3β following reperfusion (31). Nishihara et al (33) indicated a negative correlation
between tissue levels of phosphorylated GSK3β within 5 min after
reperfusion, and of infarct size within 2 h after reperfusion.
Collectively, these previous findings suggest that the
phosphorylation state of GSK3β may be associated with the degree of
myocardial reperfusion injury.
TDZD-8, a non-selective ATP-competitive inhibitor of
GSK3β, phosphorylates GSK3β at Ser9 and thus decreases the activity
and elevates the mPTP opening threshold in reperfusion injury
(44). TDZD-8 does not inhibit the
other series of kinases, such as protein kinase A and C, casein
kinase II and cyclin-dependent kinase 1. Previous results have
indicated the cardioprotective effect of TDZD-8 in myocardial
ischemia/reperfusion injury, which resulted in the phosphorylation
of GSK3βser9 (34). In the present
study, pharmacological postconditioning with 2 mg/kg atorvastatin
calcium did not further attenuate myocardial ischemia reperfusion
injury after TDZD-8 phosphorylated and inactivated GSK3β. On the
basis of these results we propose that atorvastatin has the same
method of action as TDZD-8 in the reperfusion injury in rats with
early stages of streptozotocin-induced diabetes.
It is also notable that GSK3β should be administered
at an optimum concentration level. In the present experiment, 2
mg/kg atorvastatin calcium was considered to be a relatively
optimal dose; however, more research is required, such as the
optimal atorvastatin calcium dose in the progress of alleviating
myocardial ischemia/reperfusion injury for the different species,
or at different stages of diabetes. The activity of GSK3β is
closely associated with I/R injury, and its inhibitors could reduce
the injury. However, it should be noted that the effect of the
phosphorylation of GSK3β may differ between species of organism,
and GSK3β inhibitors have a number of side effects. For example,
studies have indicated that long-term use of GSK3β inhibitors in
patients with heart failure may cause ventricular hypertrophy and
tumor growth (43). Therefore, more
comprehensive and in-depth studies are required to investigate the
most effective use of the phosphorylation or directly inhibition of
GSK3β in the context of treating I/R injury.
In the present study, immunohistochemical methods
were used to detect the distribution of p-GSK3β, and the results
suggested that p-GSK3β was distributed in the cytoplasm and
nucleus. Although p-GSK3β is inactivated, it may continue to
phosphorylate numerous proteins, including HSF-1 (50). HSF-1 is an important pro-survival
transcription factor which is induced to trimerization in the
cytoplasm after being phosphorylated, and subsequently combines
with the heat shock element HSP promoter region in the nucleus to
synthesize HSP (4,6,51–53).
HSPs are a class of endogenous proteins that act
against various types of cellular stress, including heat shock,
ischemia and hypoxia, and are highly conserved evolutionarily. HSPs
are named according to their molecular weight, including HSP90,
HSP84, HSP70, HSP60, HSP27 and HSP20. HSP90, HSP84, HSP70, HSP27
and HSP20 have been associated with cardiovascular disease
(54).
Xu et al (30)
reported that the PI3K/AKt pathway could increase the level of
HSP70, and resulted in a cytoprotective effect in human tissues.
Furthermore, other studies have suggested that HSP70 exerts a
protective effect in non-diabetic myocardium, including the
protection of myocardial contractility (51,52),
reduction of myocardial necrosis (6)
and inducing an antiarrhythmic effect (4).
It has been reported that in the PI3K/Akt pathway,
Stat3 could be the upstream signal of HSF-1, and thus promote the
synthesis of HSP70 during myocardial reperfusion (50). In the present experiment, the changes
of HSF-1 and serum HSP70 were comparable to those of p-GSK3β in
cardiomyocytes. According to the results of this experiment and
other experiments, HSF-1 could be phosphorylated by p-GSK3β, and
p-GSK3β may accelerate HSP70 production partially by activating
HSF-1 during the experimental conditions, thus serving a protective
function.
DMSO is a colorless organosulfur liquid and an
important polar aprotic solvent that dissolves both polar and
nonpolar compounds. DMSO is known to possess antioxidative,
anti-inflammatory and analgesic properties, and to promote blood
circulation. Furthermore, DMSO has been applied clinically in the
treatment of gastrointestinal diseases, rheumatism and amyloidosis
disease. On the basis of its antioxidative and anti-inflammatory
properties, numerous prior studies have observed the effects of
DMSO in various organs during myocardial ischemia, including the
heart (55), brain (56), liver (57), gastrointestinal (58), lung (59) and ovaries (60). The usage and dosage were different,
and the results vary from against reperfusion injury (3,55,61), no
protect effect (3,5,62) to
aggravate damage (63). In the
present study, TDZD-8 and atorvastatin calcium were dissolved in
0.8% DMSO for administration to rats, and indexes of cardiac injury
were compared between DM IR and DM DMSO groups. No significant
differences in the level of myocardial infarction size, cTnI,
morphological lesion of myocardial cell or the level of p-GSK3β
were detected between the DM IR and DM DMSO group. The results of
the present study therefore indicate that the 0.8% concentration of
DMSO used had no influence in the result.
Nevertheless, the present study contained a number
of limitations. First, the results of animal diabetic models in
MIRI were incomparable from one to another, reduce infarct size,
and enlarge infarct size or no change. Due to differences in
experimental design details, a single factor is insufficient to
explain the marked variance in the effects of diabetes on the
infarct size. Therefore, we compared the tolerance against
infarction between DM and NDM groups, and the model we used proved
more sensitive to myocardial ischemia. Additional estrogen usage
may result in decreased infarct size (35). Second, previous experiments suggest
that the infarct area in females are smaller than in males relative
to body mass (49,64,65). In
prior experiments (64,65), it has been suggested that additional
preconditioning and other cardioprotective approaches do not have a
further protective effect in females. This may be due to the
interactions between cardioprotective signaling pathways in female
animals and pre- and post-conditioning signaling pathways.
Considering the successful rate of diabetic model, we selected male
SD rats. Moreover, our study didn't investigate the time-and
dose-effect of atorvastatin calcium postconditioning on MIRI. On
the basis of the above evidence, the relevance of the present
results is limited to the specific settings, and cannot be
generalized. Finally, numerous pharmacological agents that show
promise in a laboratory-based animal model have not reduced the
infarct size in clinical trials as expected. Thus, atorvastatin
calcium postconditioning as presently described is far from
clinical use, and more research is required.
Pharmacological postconditioning with atorvastatin
calcium can attenuate diabetic diabetic heart ischemia/reperfusion
injury in the present settings, and the protective effect of 2
mg/kg group was improved compared with the 1 mg/kg group. The
phosphorylation of GSK3β may play a critical role in this
protective process in diabetic rats, and p-GSK3β can may induce
HSP70 expression partially by activating HSF-1 during MIRI.
Acknowledgements
The present work was supported by a grant from
Fujian Provincial Department of Education Focus on Science and
Technology Projects (grant no. JA12135). The authors would like to
thank Mrs Hong Zheng and staff from Union Hospital for assisting in
data collection, and all colleagues who helped in preparation for
the current study.
Glossary
Abbreviations
Abbreviations:
|
GSK3β
|
glycogen synthase kinase 3β
|
References
|
1
|
Jennings RB, Sommers HM, Smyth GA, Flack
HA and Linn H: Myocardial necrosis induced by temporary occlusion
of a coronary artery in the dog. Arch Pathol. 70:68–78.
1960.PubMed/NCBI
|
|
2
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moore RM, Muir WW, Bertone AL, Beard WL
and Stromberg PC: Effects of dimethyl sulfoxide, allopurinol,
21-aminosteroid U-74389G, and manganese chloride on low-flow
ischemia and reperfusion of the large colon in horses. Am J Vet
Res. 56:671–687. 1995.PubMed/NCBI
|
|
4
|
Meng X, Brown JM, Ao L, Banerjee A and
Harken AH: Norepinephrine induces cardiac heat shock protein 70 and
delayed cardioprotection in the rat through alpha 1 adrenoceptors.
Cardiovasc Res. 32:374–383. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Horne MM, Pascoe PJ, Ducharme NG, Barker
IK and Grovum WL: Attempts to modify reperfusion injury of equine
jejunal mucosa using dimethylsulfoxide, allopurinol, and
intraluminal oxygen. Vet Surg. 23:241–249. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Okubo S, Wildner O, Shah MR, Chelliah JC,
Hess ML and Kukreja RC: Gene transfer of heat-shock protein 70
reduces infarct size in vivo after ischemia/reperfusion in the
rabbit heart. Circulation. 103:877–881. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marfella R, D'Amico M, Di Filippo C,
Piegari E, Nappo F, Esposito K, Berrino L, Rossi F and Giugliano D:
Myocardial infarction in diabetic rats: role of hyperglycaemia on
infarct size and early expression of hypoxia-inducible factor 1.
Diabetologia. 45:1172–1181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F,
Wang NP, Guyton RA and Vinten-Johansen J: Inhibition of myocardial
injury by ischemic postconditioning during reperfusion: comparison
with ischemic preconditioning. Am J Physiol Heart Circ Physiol.
285:H579–H588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ravingerová T, Neckár J and Kolár F:
Ischemic tolerance of rat hearts in acute and chronic phases of
experimental diabetes. Mol Cell Biochem. 249:167–174. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kin H: Postconditioning attenuates
myocardial ischemia-reperfusion injury by inhibiting events in the
early minutes of reperfusion. Cardiovasc Res. 62:74–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marfella R, Di Filippo C, Esposito K,
Nappo F, Piegari E, Cuzzocrea S, Berrino L, Rossi F, Giugliano D
and D'Amico M: Absence of inducible nitric oxide synthase reduces
myocardial damage during ischemia reperfusion in
streptozotocin-induced hyperglycemic mice. Diabetes. 53:454–462.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Di Filippo C, Marfella R, Cuzzocrea S,
Piegari E, Petronella P, Giugliano D, Rossi F and D'Amico M:
Hyperglycemia in streptozotocin-induced diabetic rat increases
infarct size associated with low levels of myocardial HO-1 during
ischemia/reperfusion. Diabetes. 54:803–810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lowenstein CJ: Myocardial reperfusion
injury. N Engl J Med. 357:2409, 2409–2410. 2007.
|
|
14
|
Crunkhorn S: Cardiovascular drugs:
Engineered apyrase averts clot formation. Nat Rev Drug Discov.
13:724–725. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lund SS: Ischaemic conditioning for
myocardial salvage after AMI. Lancet. 375:1691–1692. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meybohm P, Bein B, Brosteanu O, Cremer J,
Gruenewald M, Stoppe C, Coburn M, Schaelte G, Böning A, Niemann B
and Roesner J: A multicenter trial of remote ischemic
preconditioning for heart surgery. N Engl J Med. 373:1397–1407.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hausenloy DJ and Yellon DM: Targeting
myocardial reperfusion injury - The search continues. N Engl J Med.
373:1073–1075. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cung TT, Morel O, Cayla G, Rioufol G,
Garcia-Dorado D, Angoulvant D, Bonnefoy-Cudraz E, Guérin P, Elbaz
M, Delarche N and Coste P: Cyclosporine before PCI in patients with
acute myocardial infarction. N Engl J Med. 373:1021–1031.
2015.PubMed/NCBI
|
|
19
|
Keeley EC, Boura JA and Grines CL: Primary
angioplasty versus intravenous thrombolytic therapy for acute
myocardial infarction: a quantitative review of 23 randomised
trials. Lancet. 361:13–20. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boston DR, Malouf A and Barry WH:
Management of intracoronary thrombosis complicating percutaneous
transluminal coronary angioplasty. Clin Cardiol. 19:536–542. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tissier R, Waintraub X, Couvreur N,
Gervais M, Bruneval P, Mandet C, Zini R, Enriquez B, Berdeaux A and
Ghaleh B: Pharmacological postconditioning with the phytoestrogen
genistein. J Mol Cell Cardiol. 42:79–87. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nicholls SJ, Brewer HB, Kastelein JJ,
Krueger KA, Wang MD, Shao M, Hu B, McErlean E and Nissen SE:
Effects of the CETP inhibitor evacetrapib administered as
monotherapy or in combination with statins on HDL and LDL
cholesterol: A randomized controlled trial. JAMA. 306:2099–2109.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jones SP, Gibson MF, Rimmer DR, Gibson TM,
Sharp BR and Lefer DJ: Direct vascular and cardioprotective effects
of rosuvastatin, a new HMG-CoA reductase inhibitor. J Am Coll
Cardiol. 40:1172–1178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Heart Protection Study Collaborative
Group, . MRC/BHF Heart Protection Study of cholesterol lowering
with simvastatin in 20,536 high-risk individuals: A randomised
placebo-controlled trial. Lancet. 360:7–22. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ueda Y, Kitakaze M, Komamura K, Minamino
T, Asanuma H, Sato H, Kuzuya T, Takeda H and Hori M: Pravastatin
restored the infarct size-limiting effect of ischemic
preconditioning blunted by hypercholesterolemia in the rabbit model
of myocardial infarction. J Am Coll Cardiol. 34:2120–2125. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ludman A, Venugopal V, Yellon DM and
Hausenloy DJ: Statins and cardioprotection - more than just lipid
lowering? Pharmacol Ther. 122:30–43. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liao JK and Laufs U: Pleiotropic effects
of statins. Annu Rev Pharmacol Toxicol. 45:89–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nahrendorf M, Sosnovik D, Chen JW, Panizzi
P, Figueiredo JL, Aikawa E, Libby P, Swirski FK and Weissleder R:
Activatable magnetic resonance imaging agent reports
myeloperoxidase activity in healing infarcts and noninvasively
detects the antiinflammatory effects of atorvastatin on
ischemia-reperfusion injury. Circulation. 117:1153–1160. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Birnbaum Y, Ye Y, Rosanio S, Tavackoli S,
Hu ZY, Schwarz ER and Uretsky BF: Prostaglandins mediate the
cardioprotective effects of atorvastatin against
ischemia-reperfusion injury. Cardiovasc Res. 65:345–355. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu G, Takashi E, Kudo M, Ishiwata T and
Naito Z: Contradictory effects of short- and long-term
hyperglycemias on ischemic injury of myocardium via intracellular
signaling pathway. Exp Mol Pathol. 76:57–65. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gomez L, Paillard M, Thibault H, Derumeaux
G and Ovize M: Inhibition of GSK3beta by postconditioning is
required to prevent opening of the mitochondrial permeability
transition pore during reperfusion. Circulation. 117:2761–2768.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bopassa JC, Michel P, Gateau-Roesch O,
Ovize M and Ferrera R: Low-pressure reperfusion alters
mitochondrial permeability transition. Am J Physiol Heart Circ
Physiol. 288:H2750–H2755. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nishihara M, Miura T, Miki T, Sakamoto J,
Tanno M, Kobayashi H, Ikeda Y, Ohori K, Takahashi A and Shimamoto
K: Erythropoietin affords additional cardioprotection to
preconditioned hearts by enhanced phosphorylation of glycogen
synthase kinase-3beta. AJP: Heart and Circulatory Physiology.
291:H748–H755. 2006.
|
|
34
|
Gao H, Yin Z, Zhou N, Feng X, Gao F and
Wang H: Glycogen synthase kinase 3 inhibition protects the heart
from acute ischemia-reperfusion injury via inhibition of
inflammation and apoptosis. J Cardiovasc Pharm. 52:286–292. 2008.
View Article : Google Scholar
|
|
35
|
Booth EA, Obeid NR and Lucchesi BR:
Activation of estrogen receptor- protects the in vivo rabbit heart
from ischemia-reperfusion injury. AJP: Heart and Circulatory
Physiology. 289:H2039–H2047. 2005.
|
|
36
|
Miki T, Itoh T, Sunaga D and Miura T:
Effects of diabetes on myocardial infarct size and cardioprotection
by preconditioning and postconditioning. Cardiovasc Diabetol.
11:672012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang T, Qiao S, Lei S, Liu Y, Ng KF, Xu A,
Lam KS, Irwin MG and Xia Z: N-acetylcysteine and allopurinol
synergistically enhance cardiac adiponectin content and reduce
myocardial reperfusion injury in diabetic rats. PLoS One.
6:e239672011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miki T, Miura T, Hotta H, Tanno M, Yano T,
Sato T, Terashima Y, Takada A, Ishikawa S and Shimamoto K:
Endoplasmic reticulum stress in diabetic hearts abolishes
erythropoietin-induced myocardial protection by impairment of
phospho-glycogen synthase kinase-3-mediated suppression of
mitochondrial permeability transition. Diabetes. 58:2863–2872.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jope RS and Johnson GVW: The glamour and
gloom of glycogen synthase kinase-3. Trends Biochem Sci. 29:95–102.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jope RS, Yuskaitis CJ and Beurel E:
Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and
therapeutics. Neurochem Res. 32:577–595. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ma G, Al-Shabrawey M, Johnson JA, Datar R,
Tawfik HE, Guo D, Caldwell RB and Caldwell RW: Protection against
myocardial ischemia/reperfusion injury by short-term diabetes:
enhancement of VEGF formation, capillary density, and activation of
cell survival signaling. Naunyn Schmiedebergs Arch Pharmacol.
373:415–427. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Embi N, Rylatt DB and Cohen P: Glycogen
synthase kinase-3 from rabbit skeletal muscle. Separation from
cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J
Biochem. 107:519–527. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cohen P and Goedert M: GSK3 inhibitors:
development and therapeutic potential. Nat Rev Drug Discov.
3:479–487. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu
Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, et al:
Glycogen synthase kinase-3beta mediates convergence of protection
signaling to inhibit the mitochondrial permeability transition
pore. J Clin Invest. 113:1535–1549. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mora A, Sakamoto K, McManus EJ and Alessi
DR: Role of the PDK1-PKB-GSK3 pathway in regulating glycogen
synthase and glucose uptake in the heart. FEBS Lett. 579:3632–3638.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sugden PH, Fuller SJ, Weiss SC and Clerk
A: Glycogen synthase kinase 3 (GSK3) in the heart: A point of
integration in hypertrophic signalling and a therapeutic target? A
critical analysis. Br J Pharmacol. 153 Suppl 1:S137–S153. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Haworth RA and Hunter DR: The Ca2+-induced
membrane transition in mitochondria. II. Nature of the Ca2+ trigger
site. Arch Biochem Biophys. 195:460–467. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Halestrap AP: A pore way to die: The role
of mitochondria in reperfusion injury and cardioprotection. Biochem
Soc Trans. 38:841–860. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lagranha CJ, Deschamps A, Aponte A,
Steenbergen C and Murphy E: Sex differences in the phosphorylation
of mitochondrial proteins result in reduced production of reactive
oxygen species and cardioprotection in females. Circ Res.
106:1681–1691. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lee KH, Jeong J and Yoo CG: Positive
feedback regulation of heat shock protein 70 (Hsp70) is mediated
through Toll-like receptor 4-PI3K/Akt-glycogen synthase
kinase-3beta pathway. Exp Cell Res. 319:88–95. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nayeem MA, Hess ML, Qian YZ, Loesser KE
and Kukreja RC: Delayed preconditioning of cultured adult rat
cardiac myocytes: role of 70- and 90-kDa heat stress proteins. Am J
Physiol. 273:H861–H868. 1997.PubMed/NCBI
|
|
52
|
Jayakumar J, Suzuki K, Khan M, Smolenski
RT, Farrell A, Latif N, Raisky O, Abunasra H, Sammut IA, Murtuza B,
et al: Gene therapy for myocardial protection: transfection of
donor hearts with heat shock protein 70 gene protects cardiac
function against ischemia-reperfusion injury. Circulation.
102:I302–I306. 2000. View Article : Google Scholar
|
|
53
|
Son TW, Yun SP, Yong MS, Seo BN, Ryu JM,
Youn HY, Oh YM and Han HJ: Netrin-1 protects hypoxia-induced
mitochondrial apoptosis through HSP27 expression via DCC- and
integrin alpha6beta4-dependent Akt, GSK-3beta, and HSF-1 in
mesenchymal stem cells. Cell Death Dis. 4:e5632013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Benjamin IJ and McMillan DR: Stress (heat
shock) proteins: molecular chaperones in cardiovascular biology and
disease. Circ Res. 83:117–132. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dmitriev YV, Minasian SM, Demchenko EA and
Galagudza MM: Cardioprotective properties of dimethyl sulfoxide
during global ischemia-reperfusion of isolated rat heart. Bull Exp
Biol Med. 154:47–50. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
de la Torre JC: Role of dimethyl sulfoxide
in prostaglandin-thromboxane and platelet systems after cerebral
ischemia. Ann N Y Acad Sci. 411:293–308. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sahin M, Avsar FM, Ozel H, Topaloglu S,
Yılmaz B, Pasaoglu H, Avunduk MC, Erikoglu M and Hengirmen S: The
effects of dimethyl sulfoxide on liver damage caused by
ischemia-reperfusion. Transplant Proc. 36:pp. 2590–2592. 2004;
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Schoenberg MH and Beger HG: Oxygen
radicals in intestinal ischemia and reperfusion. Chem Biol
Interact. 76:141–161. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Punch J, Rees R, Cashmer B, Oldham K,
Wilkins E and Smith DJ: Acute lung injury following reperfusion
after ischemia in the hind limbs of rats. J Trauma. 31:760–765,
765-767. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ergun Y, Koc A, Dolapcioglu K, Akaydin Y,
Dogruer G, Kontas T, Kozlu T and Aslan E: The protective effect of
erythropoietin and dimethylsulfoxide on ischemia-reperfusion injury
in rat ovary. Eur J Obstet Gynecol Reprod Biol. 152:186–190. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Parisi A, Alfieri A, Mazzella M, Mazzella
A, Scognamiglio M, Scognamiglio G, Mascolo N and Cicala C:
Protective effect of dimethyl sulfoxide on acute myocardial
infarction in rats. J Cardiovasc Pharmacol. 55:106–109. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Guimaraes SB, Kimura OS and Vasconcelos
PR: Dimethylsulfoxide attenuates ischemia-reperfusion injury in rat
testis. Acta Cir Bras. 25:357–361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Weinstein PR, Hameroff SR, Johnson PC and
Anderson GG: Effect of hyperbaric oxygen therapy or dimethyl
sulfoxide on cerebral ischemia in unanesthetized gerbils.
Neurosurgery. 18:528–532. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Talukder MA, Yang F, Shimokawa H and
Zweier JL: eNOS is required for acute in vivo ischemic
preconditioning of the heart: effects of ischemic duration and sex.
Am J Physiol Heart Circ Physiol. 299:H437–H445. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Shinmura K, Nagai M, Tamaki K and Bolli R:
Gender and aging do not impair opioid-induced late preconditioning
in rats. Basic Res Cardiol. 99:46–55. 2004. View Article : Google Scholar : PubMed/NCBI
|