Introduction
Bronchial remodeling involves a series of chronic
injuries in airway wall cells and repair processes characterized by
variations in the cellular composition, quantity and structure. It
mainly manifests as extracellular matrix (ECM) deposition, basement
membrane thickening, airway smooth muscle (ASM) hyperplasia and
hypertrophy (1–3). Bronchial remodeling is the pathological
basis for chronicity, sustainability, and exacerbation of bronchial
remodeling (3). It is the
pathological basis for irreversible bronchial obstruction as well
as steroid-resistant asthma (4).
Consequently, inhibition of bronchial remodeling is an attractive
target to facilitate control of the occurrence and development of
bronchial asthma.
Transglutaminase 2, also known as tissue
transglutaminase (tTG) (5), is a
member of the transglutaminase family (5,6). tTG is
a predominantly cytoplasmic protein which is also detectable in the
nucleus, plasma membrane and ECM (5). tTG can post-translationally modify ECM
proteins to form stable structures, which are resistant to
degradation, thus leading to ECM protein deposition as well as
tissue fibrosis (6–8). The content and activity of tTG protein
is markedly increased in renal interstitial fibrosis in animal
models and diseased kidney tissue samples (9). Consequently, tTG is considered an
important regulatory protein in tissue fibrosis and remodeling.
However, the specific mechanism has remained to be clarified and
requires further study.
Triggering receptor expressed on myeloid cells-1
(TREM-1) is mainly expressed in neutrophils, granulocytes and
monocytes/giant cells (10). Under
conditions of infection and inflammation, the expression levels of
TREM-1 are significantly upregulated (11). Activation of TREM-1 not only mediates
genetic transcription of pro-inflammatory cytokines, chemokine
receptors and cell surface molecules (12), but also increases the generation of
cytokines [tumor necrosis factor (TNF)-α, interleukin (IL)-lβ and
IL-6] through the synergistic activation of Toll-like receptors and
Nod-like receptors (12).
Atorvastatin is a
3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase
inhibitor. It can inhibit the transformation of HMG-CoA to
hydroxyvaleric acid (MA), the precursor of isoprene. Metabolites of
MA, including, farnesyl diphosphate (FPP) and geranylgeranyl
pyrophosphate (GGPP), not only act as cholesterol precursors
(13), but also participate in the
activation of small G-proteins of the Ras and Rho protein family.
FPP and GGPP mainly participate in the generation of inflammation
as well as cell proliferation and migration, macrophage
phagocytosis and small vessel leakage (13,14).
They also promote the release of transforming growth factor
(TGF)-β1, vascular endothelial growth factor (VEGF) and matrix
metalloproteinases (MMPs) and the expression of nitric oxide
synthase-2, and activate the nuclear factor (NF)-κB pathway
(15,16). Therefore, it is speculated that
atorvastatin may inhibit bronchial remodeling in bronchial asthma
in mice.
The present study found that atorvastatin inhibited
the expression of tTG, reduced the activity of Smad3/TGF-β and
collagen deposition, and improved the remodeling of lung tissue in
mice with bronchial asthma. Atorvastatin reduced the expression of
NF-κB p65, increased the expression of nuclear factor erythroid
2-related factor (Nrf)2, and reduced tracheal inflammation and
peroxidative reactions through inhibition of the expression of
TREM-1. It also decreased the expression of MMP-9 and TGF-β1, and
inhibited lung tissue remodeling in mice with bronchial asthma.
Materials and methods
Chemicals and reagents
Ovalbumin (OVA; grade V) and dexamethasone (Dex)
were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Atorvastatin was purchased from Pfizer (New York City, NY, USA).
Aluminum hydroxide was obtained from Pierce (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The TGF-β1 ELISA kit was from
R&D Systems (Minneapolis, MN, USA).
Animals
Six-week-old female BALB/c mice were purchased from
the Kunming Medical University Animal Center (Kunming, China) and
bred in-house. All experimental procedures were approved by the
Animal Care and Use Committee of Kunming Medical University. The
experimental procedures were approved by the Ethics Committee of
Kunming Medical University (Kunming, China).
Murine model of chronic asthma
The model was established according to a protocol by
Jain et al (17). In brief,
mice were sensitized by means of intraperitoneal injection of OVA
(10 µg) precipitated with aluminum hydroxide (100 µg) on days 0 and
14. Subsequently, mice were given 1.5 mg/kg atorvastatin
(atorvastatin group or treatment group) by oral gavage, 10 mg/kg
Dex (Dex group) or 0.2 ml phosphate-buffered saline (PBS) by means
of intraperitoneal injection 0.5 h prior to each OVA challenge
(nebulized 2.5% solution 30 min/day, 3 days/week for 6 weeks). Mice
were sacrificed 24 h after the final OVA inhalation. Control groups
were intraperitoneally injected with PBS and inhaled aerosolized
PBS at the same time as the other three groups. Mice treated with
atorvastatin or Dex but without OVA sensitization were also set up
as controls. Serum was collected on day 21 and airway
hyperresponsiveness (AHR) [enhanced pause (Penh)] to increasing
concentrations of methacholine (MCh; 1.5–12 mg/ml; Hubei Dongshang
Chemical Co., Ltd., Hubei, China) was measured by whole-body
plethysmography (Buxco Research Systems, Wilmington, NC, USA) on
day 22 (17). Lungs were used either
for bronchoalveolar lavages (BALs) and protein extraction or for
histological analyses and RNA extraction.
Extraction of RNA
For the isolation of RNA from lung tissue, mice were
sacrificed via an overdose of sodium pentobarbital (intravenous
injection, 150 mg/kg; Wuhan Dinghui Chemical Co., Ltd, Wuhan,
China) and under aseptic conditions the lung tissues were removed
and immediately frozen in liquid nitrogen. Prior to RNA extraction,
lung samples were homogenized in TRIzol reagent (Thermo Fisher
Scientific, Inc.) using a Mixer 301 (Invitrogen; Thermo Fisher
Scientific, Inc.). Total RNA was extracted according to the
manufacturer's instructions. RNA samples were electrophoresed in
agarose gel (Shanghai Yuanye Biochemicals Ltd. Shanghai, China) and
the gel image visualized using Kodak 1D software (Life
Technologies, Grand Island, NY, USA), with ethidium bromide
(Beijing Xin Hua Luyuan Science and Technology Co., Ltd, Beijing,
China) for quality control.
Reverse-transcription
semi-quantitative polymerase chain reaction analysis
(RT-semi-qPCR)
Three micrograms of RNA were reverse-transcribed
with 200 U Moloney Murine Leukemia Virus reverse transcriptase
(Shanghai Jiang Lai Biotechnology Co., Ltd., Shanghai, China) for 1
h at 37°C for synthesis of complementary (c)DNA. Quantitative
changes in mRNA expression were assessed by semi-qPCR in a Bio-Rad
CFX (Bio-Rad Laboratories, Inc., Hercules, CA, USA) using SYBR
Green PCR Master Mix (Aria-tous, Isfahan, Iran). The PCR reaction
mix was made up by 0.5 U of Taq polymerase, 2 µl of each primer and
3 µl of each cDNA sample in a final volume of 20 µl. Conditions for
amplification were 1 cycle at 94°C for 5 min followed by 40 cycles
of 94°C for 30 sec, 58°C for 30 sec and 70°C for 45 sec. All
amplifications were repeated three times. Oligonucleotide primer
sequences (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) are
listed in Table I. β-actin was used
as an endogenous control. The mRNA levels of various genes were
quantified using SYBR premix Ex Taq (Takara Bio, Inc., Otsu,
Japan), and analysis was performed using an ABI PRISM 7300 RT-PCR
System (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Ordinary PCR products were separated on a 2% agarose gel.
Expressions were normalized to endogenous controls. Relative
quantification of mRNA expression levels of target genes was
calculated using the 2−ΔΔCq method (18).
 | Table I.Primer sequences used for polymerase
chain reaction. |
Table I.
Primer sequences used for polymerase
chain reaction.
| Gene | Primer | Product length
(bp) |
|---|
| tTG |
F-5′-CAAGAACAAGGCAGACTTATCGC-3′ | 400 |
|
|
R-5′-TCTGATTATCTCGCACCAGGAAG-3′ |
|
| TGF-β1 |
F-5′-CTGCTGACCCCCACTGATAC-3′ | 298 |
|
|
R-5′-CTGTATTCCGTCTCCTTGGTTC-3′ |
|
| MMP-9 |
F-5′-GCAGGAGAGGAAGCTGAGCT-3′ | 435 |
|
|
R-5′-TCATGGTGAGAACCGAAGC-3′ |
|
| TIMP-1 |
F-5′-TCCCCAGAAATCATCGAGAC-3′ | 329 |
|
|
R-5′-ATGGCTGAACAGGGAAACAC-3′ |
|
| β-actin |
F-5′-GCCATGTACGTAGCATCCA-3′ | 374 |
|
|
R-5′-GAACCGCTCATTGCCGATAG-3′ |
|
Western blot analysis
Lung tissues were homogenized in lysis buffer
containing protease inhibitors and protein concentrations were
determined using the Bradford method (Bio-Rad Laboratories Inc.). A
total of 50 µg protein from each sample was subjected to 15%
SDS-PAGE at 120 V for 90 min and separated proteins were
transferred onto polyvinylidene difluoride membranes (GE
Healthcare, Little Chalfont, UK) by the wet transfer method (250
mA, 90 min). Nonspecific sites were blocked with 5% non-fat dry
milk in Tris-buffered saline with Tween-20 (25 mM Tris, pH 7.5, 150
mM NaCl, 0.1% Tween-20) for 1 h, and the blots were then incubated
overnight at 4°C with anti-TGF-β1 antibody (1:2,000; cat. no.
sc-47778 B; Cell Signaling Technology Inc., Beverly, MA, USA),
anti-Nrf2 antibody (1:2,000; cat. no. ab137550; Sigma-Aldrich;
Merck KGaA), anti-NADPH quinine oxidoreductase (NQO1) antibody
(1:1,000; cat. no. AF7567; R&D Systems, Inc., Minneapolis, MN,
USA), anti-TREM-1 antibodies (1:1,000; cat. no. 3672; Santa Cruz
Biotechnology, Inc.), anti-VEGF antibody (1:2,000; cat. no. sc-152;
Santa Cruz Biotechnology, Inc.), anti-hypoxia-inducible factor
(HIF)-1α antibody (1:2,000; cat. no. ab114977; Serotec Ltd.,
Oxford, UK), anti-phosphorylated inhibitor of NF-κB-α (p-IκB-α;
1:1,000; cat. no. 2859; Cell Signaling Technology, Inc.),
anti-NF-κB p65 (1:1,000; cat. no. 9936; Cell Signaling Technology,
Inc.) anti-tTG antibody (1:2,000; cat. no. SG203351; BD
Biosciences, Franklin Lakes, NJ, USA) and anti-phosphorylated
(p)-Smad3 (1:2,000; cat. no. sc-130218; Santa Cruz Biotechnology,
Inc.). Subsequently, samples were incubated for 2 h at room
temperature with anti-rabbit horseradish peroxidase
(HRP)-conjugated immunoglobulin G (IgG; 1:2,000; cat. no. sc-2749;
Santa Cruz Biotechnology, Inc.) or anti-mouse HRP-conjugated IgG
(1:4,000; cat. no. 7076; Amersham Pharmacia Biotech, Piscataway,
NJ, USA) to detect binding of antibodies. The membranes were
stripped and reblotted with an anti-actin antibody (1:2,000; cat.
no. A2228; Sigma-Aldrich; Merck KGaA) to verify equal loading of
protein in each lane. The binding of the specific antibodies was
visualized by exposing to photographic film (SynGene; Synoptics
Ltd., Cambridge, UK) after treatment with an
electrochemiluminescence detection reagent (Wellstat M1M; cat. no.
310806; PerkinElmer, Inc., Waltham, MA, USA).
Immunohistochemical detection of
α-SMA
Lungs were inflation fixed at a constant pressure
(25 cm H2O) by tracheal installation of 4%
paraformaldehyde, transferred to 70% ethanol after 24 h and
embedded in paraffin as previously described (19). Immunostaining was performed on lung
sections after antigen retrieval using Retrievagen A (Zymed, San
Francisco, CA, USA) at 100°C for 20 min, and quenching endogenous
peroxidases with 3% H2O2. Sections were
blocked with 2% bovine serum albumin in PBS, followed by staining
with primary anti-α-SMA (1:2,000; cat. no. 11475; BD Pharmingen,
San Jose, CA, USA) at room temperature for 1 h. Sections were
washed and after application of the goat HRP-conjugated anti-rabbit
IgG secondary antibody (1:4,000; cat. no. HAF109; R&D Systems),
tissues were developed using Vectastain ABC (Vector Labs,
Burlingame, CA, USA) and 3,3′-diaminobenzidine (Vector Labs). After
staining, five high-power fields (magnification, ×400) were
randomly selected in each slide, and the average proportion of
cells with positive expression in each field was counted using the
true color multi-functional cell image analysis management system
(Beckman Coulter, Inc, Fullerton, CA, USA), with values expressed
as positive units (pu).
Measurement of AHR
AHR to MCh was determined on day 33 by
single-chamber body plethysmography as described previously
(20). In brief, each mouse was
placed in the Buxco single chamber, acclimatized for 5–10 min and
baseline recordings were made for 5 min. The mice were then
challenged with PBS or various concentrations of MCh aerosol and
signals were recorded with in-built software (Buxco) to determine
the Penh values, which are reliable in BALB/c mice (20). MCh PC200, which is the partial
concentration of MCh required to double the baseline Penh, Penh0,
was then calculated. To confirm the findings of the non-invasive
body plethysmography, respiratory mechanics were determined during
mechanical ventilation (FlexiVent System; Scireq, Montréal, Canada)
as previously described (20). In
brief, following anesthetisia via intraperitoneal administration of
sodium pentobarbital (90 mg/kg; Wuhan Dinghui Chemical Co., Ltd.)
mice were intubated after tracheostomy, ventilated with a
computer-controlled ventilator, and airway resistance with various
concentrations of MCh was estimated using the flexiVent system
(Scireq) that integrates the ventilator with the respiratory
mechanics.
ELISA
At 24 h after the last challenge, bronchoalveolar
lavage fluid (BALF) was obtained from the mice under anaesthesia
with intravenous injection of sodium pentobarbital (60 mg/kg),
using 1 ml sterile isotonic saline. Lavage was performed four times
in each mouse and the total volume was collected separately. The
lavage fluid sample was immediately centrifuged at 2,000 × g for 10
min at room temperature and stored at −80°C until use. The
concentrations of IL-8, IL-13, IL-17, TNF-α, MMP-9 and TIMP-1 in
BALF supernatants were assayed by ELISA (mouse IL-8 ELISA kit, cat.
no. 20760, Bio-Medical Assay Co., Ltd., Beijing, China; mouse IL-13
ELISA kit, cat. no. KMC2222; mouse IL-17 ELISA kit, cat. no.
KMC3022; mouse TNF-α ELISA kit, cat. no. KMC3011, all Thermo Fisher
Scientific, Inc.; mouse MMP-9 ELISA kit, cat. no. M0238,
Bio-Medical Assay Co., Ltd.; mouse TIMP-1 ELISA kit, cat. no.
EMTIMP1CL, Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions.
Inflammatory cell counts in BALF
Following OVA challenge of mice, BALF samples were
obtained and processed, and inflammatory cells were counted as
previously described (21). In
brief, at 48 h after the last challenge, mice were given an
intraperitoneal injection of pentobarbital (50 mg/kg; Hanlim Pharm.
Co., Seoul, Korea) and a tracheostomy was performed. To obtain
BALF, ice-cold PBS (0.6 ml) was infused into the lung and withdrawn
via tracheal cannulation three times (total volume, 1.8 ml). Total
inflammatory cell numbers were assessed by counting cells in at
least five squares of a hemocytometer after exclusion of dead cells
by Trypan blue staining. To determine differential cell counts, 100
ml of BALF was centrifuged onto slides (200 × g, 4°C, 10 min) using
a Cytospin (Hanil Science Industrial, Seoul, Korea). After slides
were dried, cells were fixed and stained using DiffQuikH staining
reagent (B4132-1A; IMEB Inc., Deerfield, IL, USA), according to the
manufacturer's instructions. The supernatant of the BALF was stored
at 27°C for cytokine measurements.
Measurement of intracellular reactive
oxygen species (ROS)
ROS were measured as previously described (22). BALF cells were washed with PBS. To
measure intracellular ROS, cells were incubated for 10 min at room
temperature with PBS containing 3.3 µM 2′,7′-dichlorofluorescein
(DCF) diacetate (Molecular Probes, Eugene, OR, USA), to label
intracellular ROS. DCF-stained cells (1×104) were
subjected to fluorescence-activated cell sorting analysis to
measure ROS levels using a FACSCalibur instrument (BD Biosciences,
San Jose, CA, USA). The data were analyzed with CellQuest Pro 3.3
software (BD Biosciences).
Measurement of glutathione (GSH) in
lung tissues
Lung tissues were homogenized with 10 ml ice-cold
lysis buffer (50 mM phosphate buffer containing 1 mM ethylene
diamine tetraacetic acid) per gram tissue. After centrifugation at
10,000 × g for 15 min at 4°C, the supernatants were removed,
deproteinated and then stored at −20°C until the samples were
assayed. Total GSH and GSH disulfide levels were determined using a
GSH Assay kit (Cayman Chemical Co., Ann Arbor, MI, USA) according
to the manufacturer's protocol.
Collagen analysis
Collagen content was measured in lung tissue
homogenates by a biochemical assay according to the manufacturer's
instructions (Sircol collagen assay, Biocolor, Carrickfergus,
Northern Ireland). Lung tissue (100 mg) was homogenized in 1 ml
Tris buffer containing 1 M sodium chloride and a protease inhibitor
cocktail (Sigma-Aldrich; Merck-Millipore KGaA, Darmstadt, Germany).
Samples were incubated overnight at 4°C with stirring and then
centrifuged, and the supernatant was assayed (23).
Hydroxyproline (Hyp)
determination
As an indirect measure of tissue collagen content,
Hyp levels in lung tissue (100 mg) were determined according to a
modified method described by Jamall et al (24). The Hyp content was expressed in
micrograms of Hyp per gram of wet weight (µg/g). The number of Hyp
measurements was the same as the number of animals in each
group.
Histopathology
After lavage, lungs were removed and fixed in
paraformaldehyde, and tissue blocks were embedded in paraffin. Lung
sections underwent deparaffinization and hydration for staining.
Hematoxylin and eosin (H&E) staining was used to evaluate ASM
thickness and the airway wall area (25). Sections were stained with Masson
Trichrome stain to evaluate collagen deposition. Images of airway
sections were randomly obtained at a magnification of ×20 with an
RT Color Digital camera (Diagnostic Instruments, Sterling Heights,
MI, USA). Airways with a ratio of maximum to minimum internal
diameter of no less than 2 were considered as cut obliquely and
were not included (25,26). In brief, five images were analyzed
per section by using Image Pro-Plus software 6.0 (Media
Cybernetics, Rockville, MD, USA) calibrated with a reference
micrometer slide. For airway wall area measurement, the basement
membrane perimeter (Pbm) and total wall area (WAt) were measured at
a magnification of ×400. The airway wall area was normalized to Pi
(WAt/Pbm) (26). ASM thickness in
H&E-stained lung sections was also evaluated by measuring the
thickness of the smooth-muscle cell layer beneath the airway
epithelial cell basement membrane (22). Collagen deposition was expressed as
the area of Masson staining per micron length of the bronchiolar
basement membrane.
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. Statistical calculations were performed using GraphPad
Prism 6.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
Significant differences among groups were identified by analysis of
variance and Tukey's honest post-hoc test was used for individual
comparisons. When variances were unequal, Welch's correction was
applied. When only two groups were compared, a one- or two-tailed
Student's t-test was applied as appropriate. P<0.05 was
considered to indicate a statistically significant difference.
Results
Atorvastatin blocks the expression of
tTG, MMP-9 and TGF-β1 in the lungs of OVA-induced asthmatic
mice
The expression levels of tTG, TIMP-1, TGF-β1 and
MMP-9 were determined in atorvastatin-treated OVA-induced asthmatic
mice by RT-semi-qPCR. As shown in Fig.
1, the gene expression levels of tTG, TIMP-1, TGF-β1, and MMP-9
in lung tissues were markedly increased after OVA inhalation
compared with the levels in the control mice. By contrast, the gene
expression levels of tTG, TIMP-1, TGF-β1 and MMP-9 in lung tissue
were markedly suppressed by the treatment with atorvastatin and
Dex. However, TIMP-1 mRNA expression in lung tissues was not
significantly changed. No significant difference was observed
between the atorvastatin group and the Dex group (P>0.05).
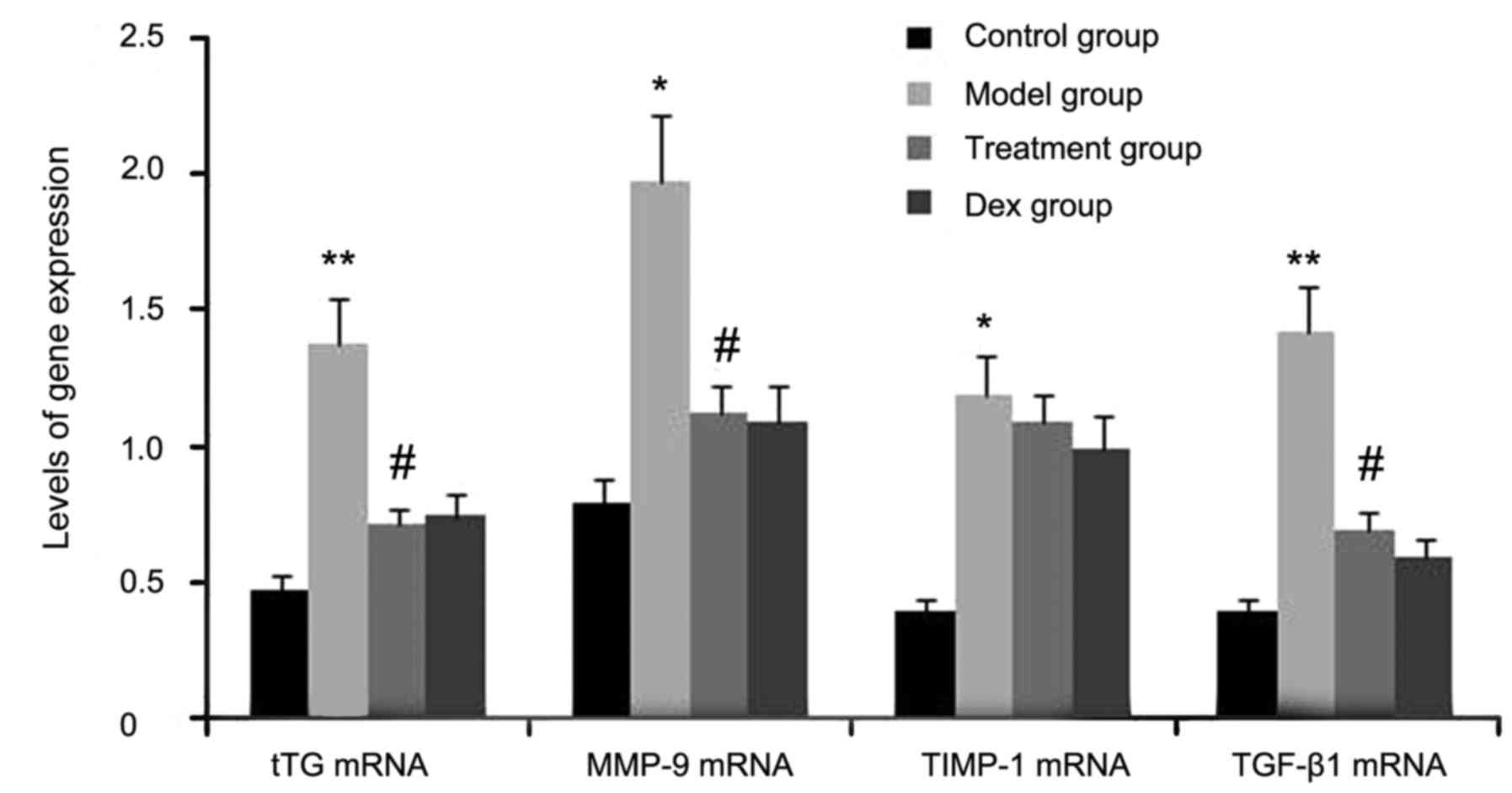 | Figure 1.Effects of atorvastatin treatment on
tTG, TIMP-1, TGF-β1 and MMP-9 gene expression in the lung tissues
of asthmatic mice. Mice were treated with atorvastatin or Dex and
challenged with OVA (nebulized 2.5% solution 30 min/day, 3
days/week for 6 weeks). The mRNA levels of tTG, TIMP-1, TGF-β1 and
MMP-9 were determined by reverse-transcription quantitative PCR
analysis. Values are expressed as the mean ± standard deviation of
one experiment consisting of three replicates. *P<0.05,
**P<0.01, vs. control group. #P<0.05 vs. model
group. PCR, polymerase chain reaction; tTG, tissue
transglutaminase; MMP-9, matrix metalloproteinase 9; TGF-β,
transforming growth factor β; TIMP-1, tissue inhibitors of
metalloproteinases 1; Dex, dexamethasone. |
Atorvastatin decreases TGF-β1, VEGF
and MMP-9 in the BALF of OVA-induced asthmatic mice
TGF-β1, VEGF, TIMP-1 and MMP-9 have a major role in
airway remodeling in asthma; therefore, their levels in the BALF
were assayed. As shown in Fig. 2,
chronic OVA challenge induced a significant increase in the BALF
levels of TGF-β1, MMP-9, VEGF and TIMP-1. This increase was
significantly reduced by atorvastatin treatment. However, TIMP-1
levels in the BALF were not significantly changed. No significant
difference was observed between atorvastatin and Dex treatment in
OVA-challenged mice.
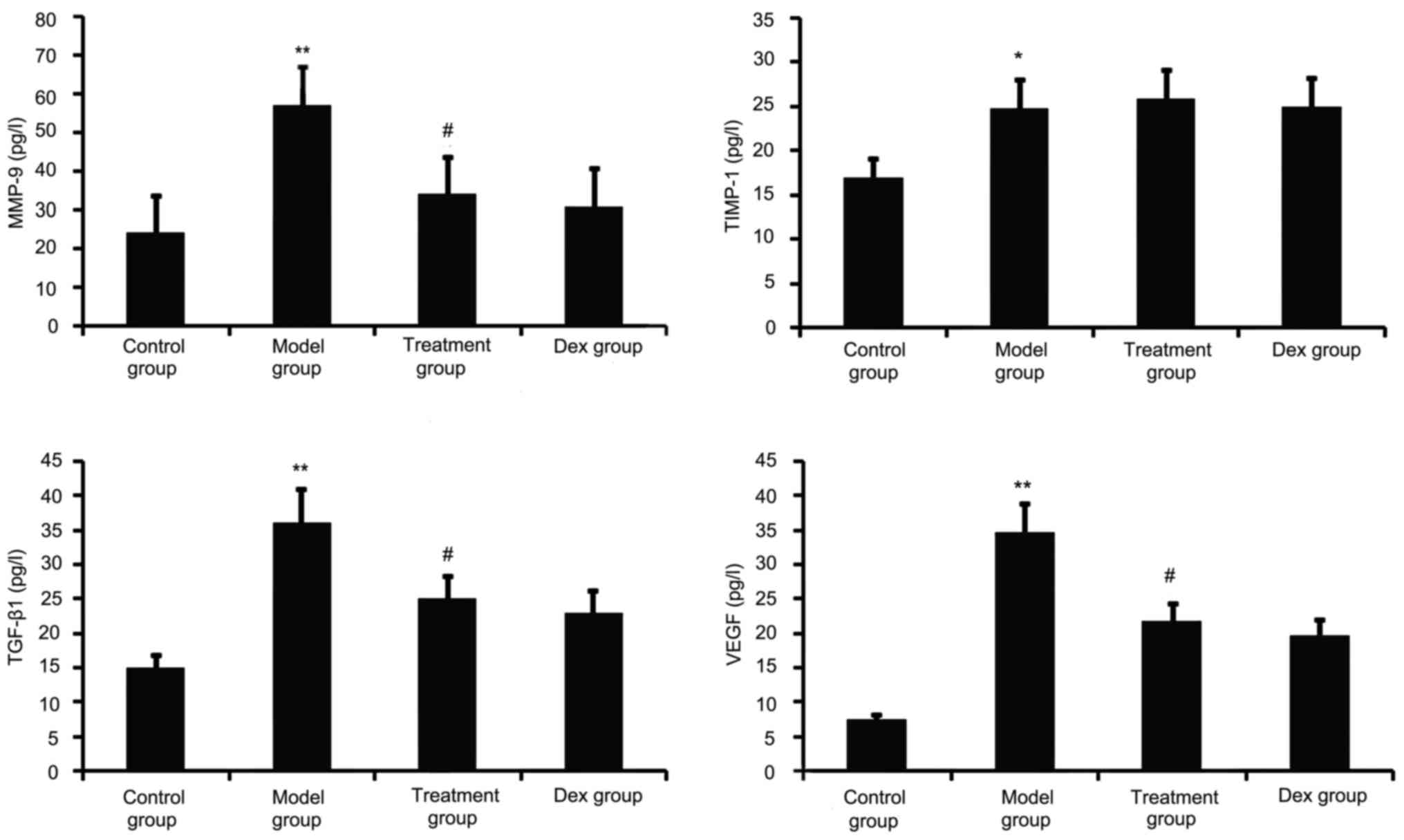 | Figure 2.Effects of atorvastatin treatment on
the levels of TGF-β1, VEGF, MMP-9 and TIMP-1 in the BALF of
asthmatic mice. Mice were treated with atorvastatin or Dex and
challenged with OVA (nebulized 2.5% solution 30 min/day, 3
days/week for 6 weeks) and the BALF levels of TGF-β1, VEGF, MMP-9
and TIMP-1 proteins were measured by ELISA. Values are expressed as
the mean ± standard error of the mean. *P<0.05, **P<0.01 vs.
control group; #P<0.05 vs. model group. MMP-9, matrix
metalloproteinase 9; VEGF, vascular endothelial growth factor;
TGF-β, transforming growth factor β; TIMP-1, tissue inhibitors of
metalloproteinases 1; Dex, dexamethasone; BALF, bronchoalveolar
lavage fluid. |
Atorvastatin inhibits TREM-2, NF-κB
p65, and p-IκB-α protein expression in OVA-induced asthmatic mouse
lungs
The expression levels of TREM-2, NF-κB p65, and
p-IκB-α protein were determined by western blot analysis. As shown
in Fig. 3, the levels of TREM-2,
NF-κB p65 and p-IκB-α protein in lung tissues were markedly
increased after OVA inhalation compared with those in the control
mice. By contrast, the levels of TREM-2, NF-κB p65 and p-IκB-α
protein in lung tissue were markedly inhibited by treatment with
atorvastatin and Dex. However, no significant difference was
observed between the atorvastatin group and the Dex group
(P>0.05).
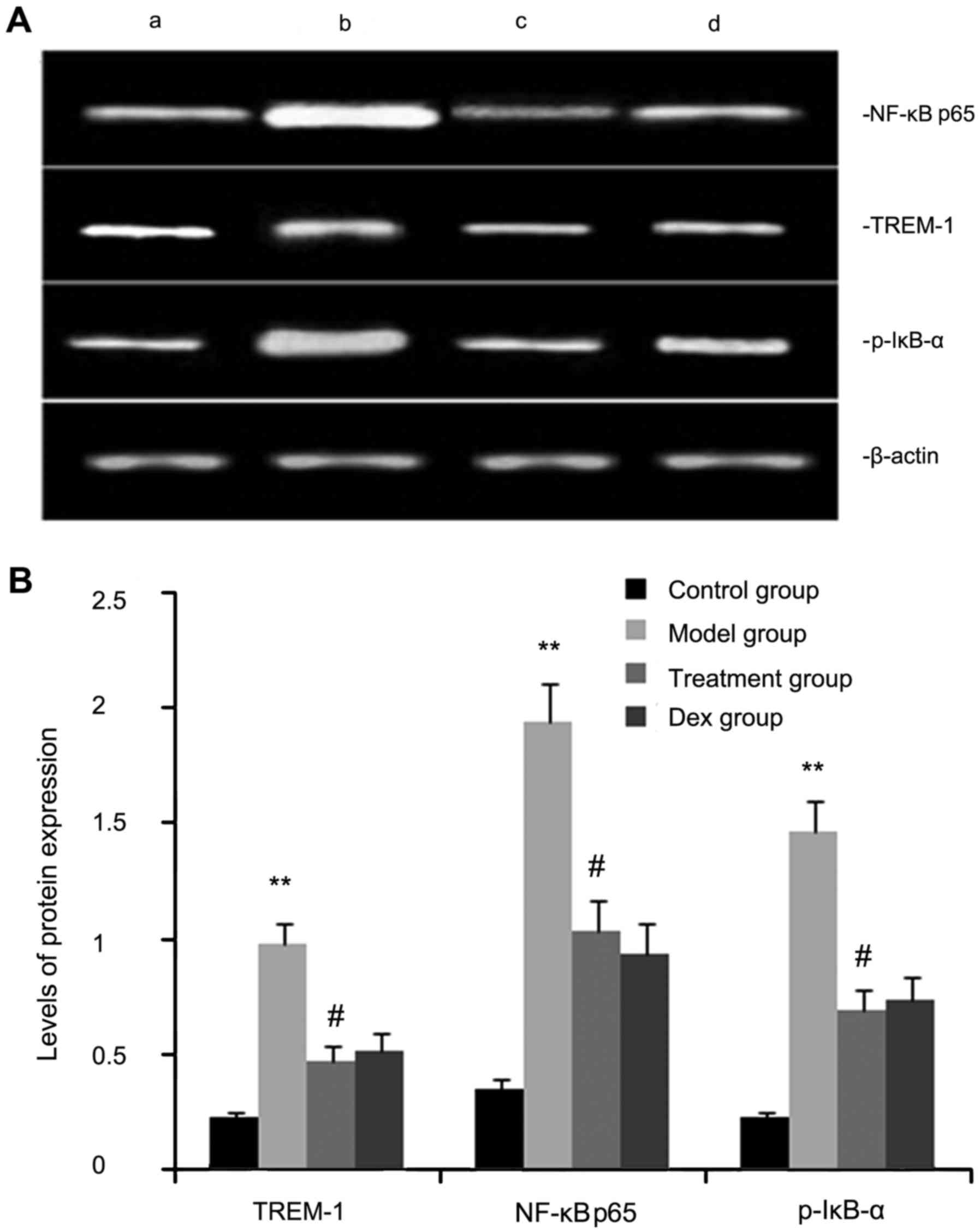 | Figure 3.Effects of atorvastatin treatment on
TREM-2, NF-κB p65 and p-IκB-α protein in the lung tissues of
asthmatic mice. Mice were treated with atorvastatin or Dex and
challenged with ovalbumin (nebulized 2.5% solution 30 min/day, 3
days/week for 6 weeks), and TREM-2, NF-κB p65 and p-IκB-α levels
were determined by western blot analysis. (A) Representative
western blot image showing the levels of TREM-2, NF-κB p65 and
p-IκB-α in the four groups of mice. Lanes: a, control group; b,
model group; c, treatment group; d, Dex group. (B)
Densitometrically quantified protein expression levels of TREM-2,
NF-κB p65 and p-IκB-α in the four groups of mice. Values are
expressed as the mean ± standard error of the mean of one
experiment consisting of three replicates. The experiments were
performed in triplicate. **P<0.01 vs. control group.
#P<0.05 vs. model group. NF-κB, nuclear factor kappa
B; p-IκB-α, phosphorylated inhibitor of NF-κB alpha; Dex,
dexamethasone; TREM-1, triggering receptor expressed on myeloid
cells 1. |
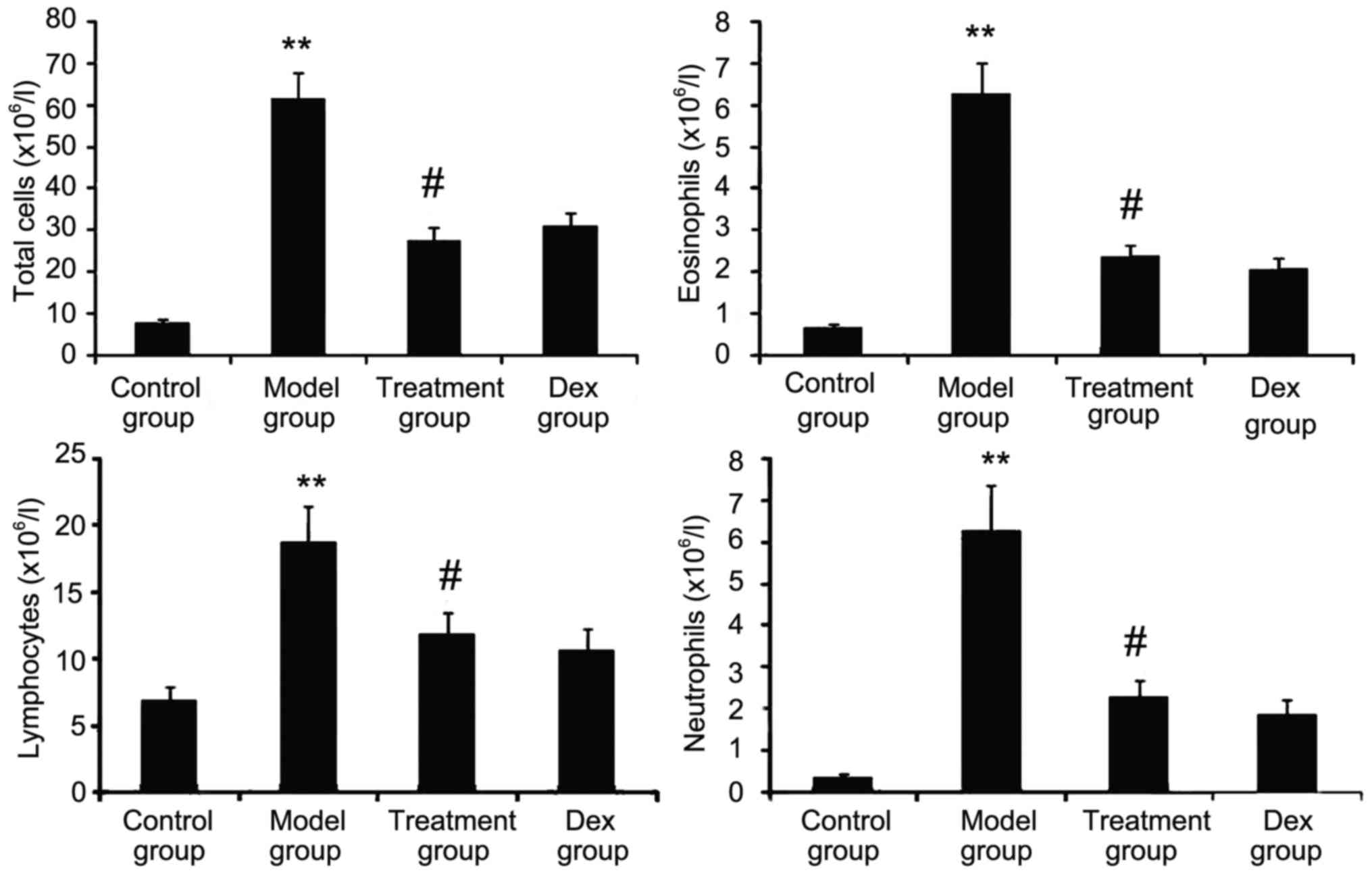
Atorvastatin prevents inflammatory
cell infiltration
The inflammatory nature of asthma is characterized
by eosinophilia and can be visualized in the OVA-challenged lung.
To determine whether atorvastatin reduces eosinophil infiltration
and alleviates lung inflammation, the effect of atorvastatin
treatment on overall lung inflammation was evaluated in
OVA-challenged mice. Compared with that in PBS-challenged mice, the
number of inflammatory cells in the BALF of OVA-challenged mice was
found to be significantly increased (Fig. 4). Furthermore, atorvastatin
administration significantly reduced the inflammatory cell
recruitment and the number of eosinophils in OVA-challenged mice.
No significant difference was observed between atorvastatin and Dex
treatments in OVA-challenged mice.
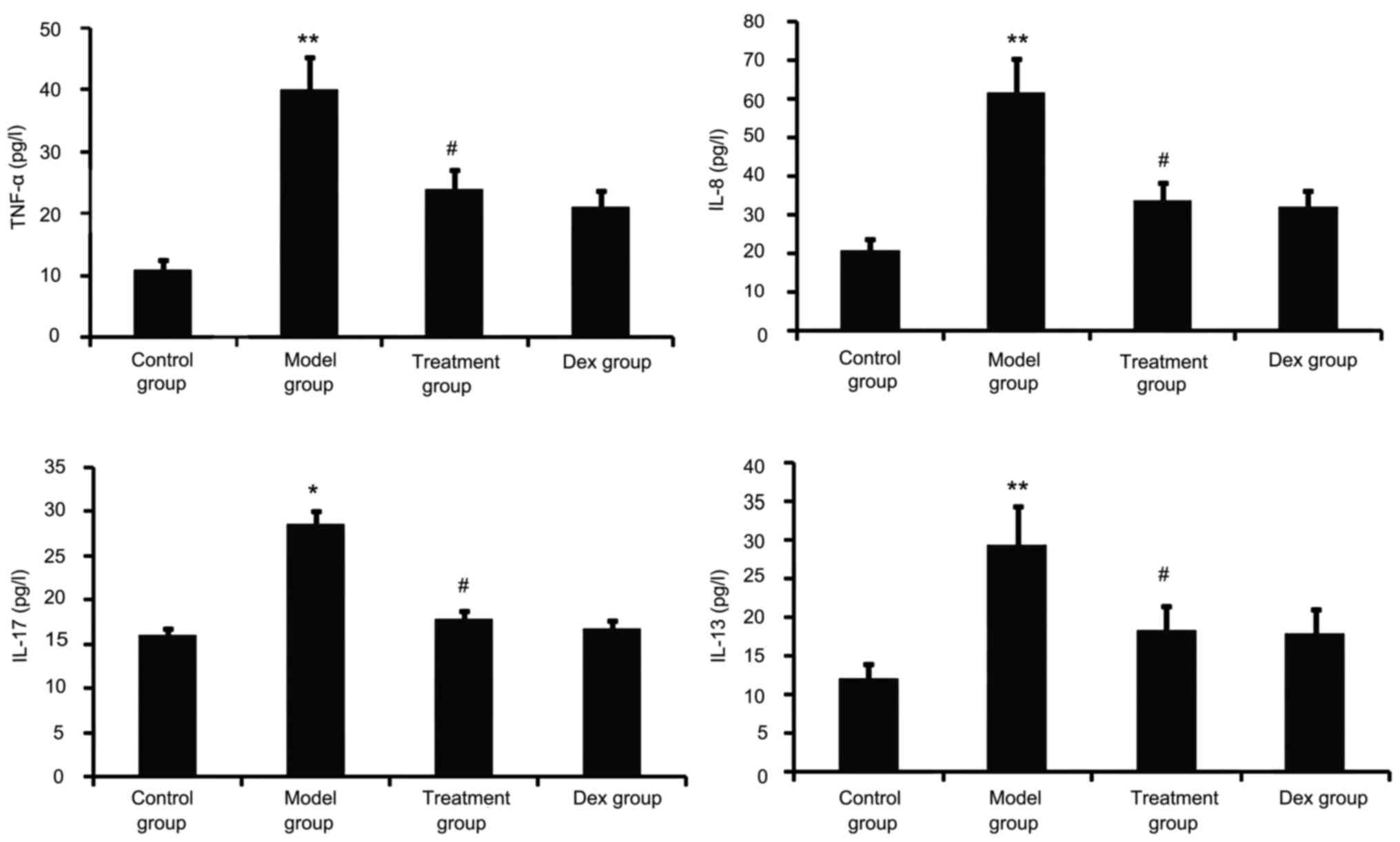
Atorvastatin blocks lung
inflammation
Airway tissue remodeling is thought to result from
chronic repetitive injury to the airway wall caused by airway
inflammation. As shown in Fig. 5,
OVA challenge increased inflammatory mediators such as TNF-α, IL-8,
IL-13 and IL-17 in the BALF. However, this was significantly
reduced in the atorvastatin and Dex treatment groups. No
significant difference was observed between the atorvastatin group
and the Dex group (P>0.05).
Effects of atorvastatin on tTG,
p-Smad3, HIF-1α, VEGF and TGF-β1 protein expression
The expression levels of tTG, p-Smad3, HIF-1α, VEGF
and TGF-β1 protein in mouse pulmonary tissue were evaluated by
western blot analysis. As shown in Fig.
6, compared with the control group, the OVA-challenged group
showed increases in tTG, p-Smad3, HIF-1α, VEGF and TGF-β1 protein
levels, which were inhibited by atorvastatin. However, no
significant difference was observed between the atorvastatin group
and the Dex group (P>0.05).
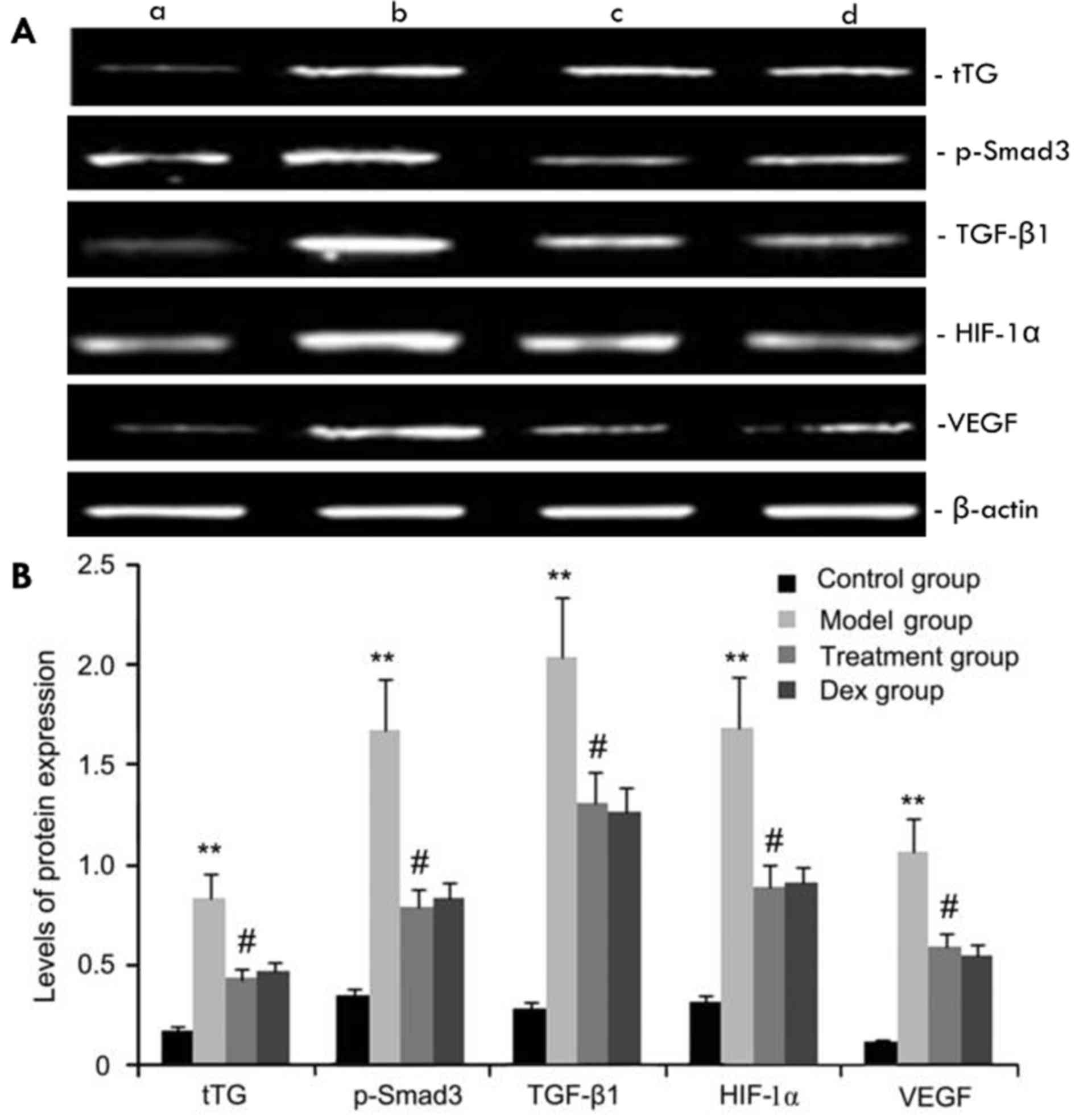 | Figure 6.Effects of atorvastatin treatment on
tTG, p-Smad3, HIF-1a, VEGF, and TGF-β1 protein expression. Mice
were treated with atorvastatin or Dex and challenged with OVA
(nebulized 2.5% solution 30 min/day, 3 days/week for 6 weeks). tTG,
p-Smad3, HIF-1α, VEGF and TGF-β1 protein were measured by western
blot analysis. (A) Representative western blots showing the protein
expression levels of tTG, p-Smad3, HIF-1α, VEGF and TGF-β1 in the
four groups of mice. Lanes: a, control group; b, model group; c,
treatment group; d, Dex group. (B) Densitometrically quantified
protein levels of tTG, p-Smad3, HIF-1α, VEGF and TGF-β1 in the four
groups. Values are expressed as the mean ± standard deviation of
one experiment consisting of three replicates. The experiments were
performed in triplicates. **P<0.05, vs. control group;
#P<0.05 vs. model group. Dex, dexamethasone; VEGF,
vascular endothelial growth factor; TGF-β, transforming growth
factor β; HIF, hypoxia-inducible factor; tTG, tissue
transglutaminase; p-Smad3, phosphorylated Smad3. |
Atorvastatin upregulates Nrf2 and NQO1
expression, decreases ROS generation, and increases GSH levels in
the lung
Next, the levels of Nrf2 and NQO1 expression, ROS
generation and GSH levels we determined in the mouse model of
chronic allergic airway disease. As shown in Fig. 7, in OVA-treated animals, Nrf2 and
NQO1 expression levels in lung tissue were significantly decreased,
ROS activity was significantly increased and GSH activity was
significantly decreased at 48 h after the last OVA inhalation
compared with the levels in the control mice. Administration of
atorvastatin and Dex significantly increased Nrf2 and NQO1
expression levels in lung tissues, inhibited ROS generation and
increased GSH activity. However, no significant difference was
observed between the atorvastatin group and the Dex group
(P>0.05).
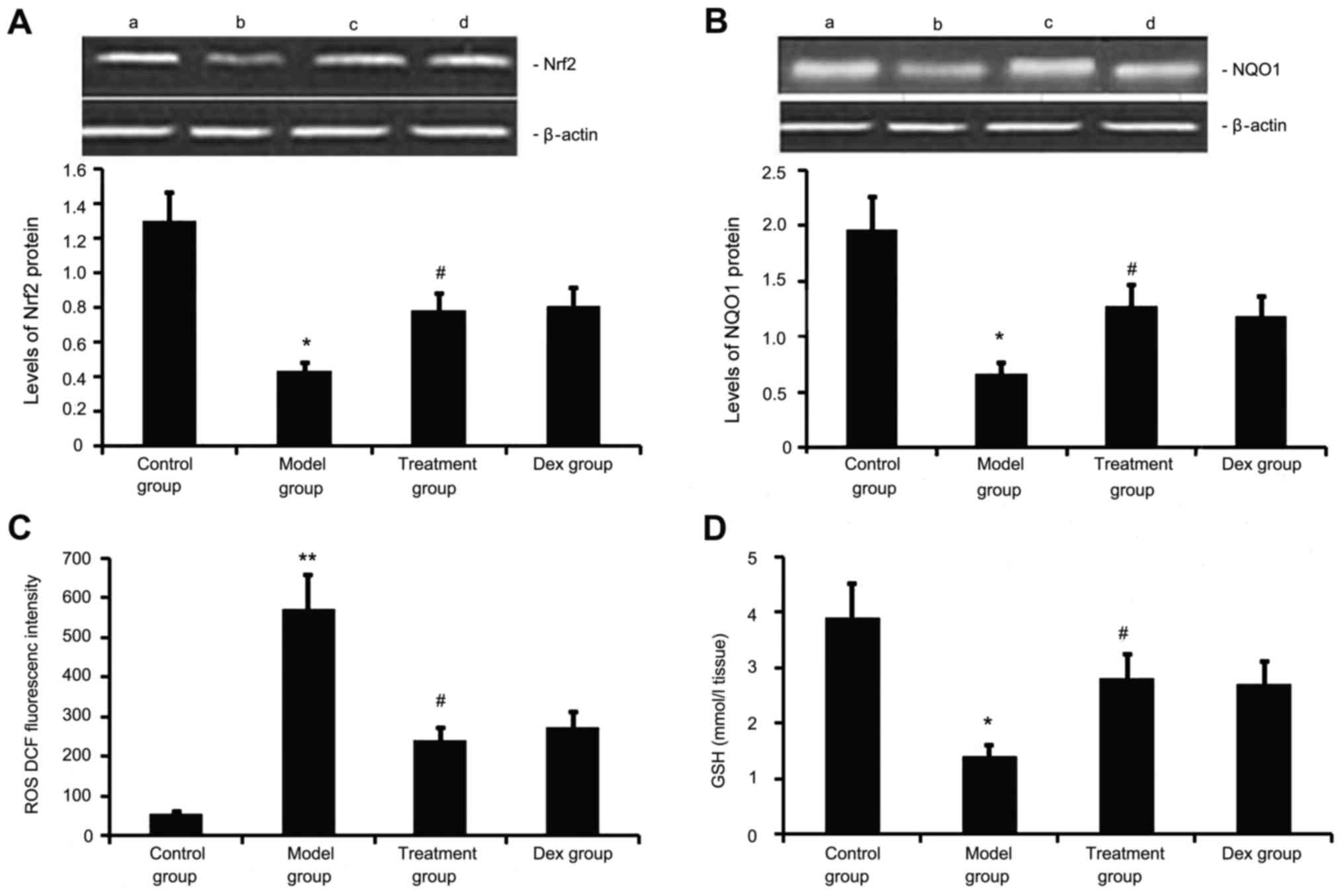 | Figure 7.Effects of atorvastatin treatment on
Nrf2 and NQO1 expression as well as ROS and GSH activities in the
lung tissues of asthmatic mice. Mice were treated with atorvastatin
or Dex and challenged with ovalbumin (nebulized 2.5% solution 30
min/day, 3 days/week for 6 weeks). The expression of (A) Nrf2 and
(B) NQO1 protein was measured by western blot analysis.
Representative western blots showing the protein expression levels
of Nrf2 and NQO1 in the four groups of mice are shown in the upper
panel. Lanes: a, control group; b, model group; c, treatment group;
d, DEX group. Densitometrically quantified protein expression
levels are shown in the lower panel. The activities of (C) ROS and
(D) GSH in lung tissues were also measured. Values are expressed as
the mean ± standard deviation of one experiment consisting of three
replicates. The experiments were performed in triplicate.
*P<0.05, **P<0.01 vs. control group; #P<0.05
vs. model group. NQO1, NADPH quinine oxidoreductase; Nrf2, nuclear
factor erythroid 2-related factor 2; ROS, reactive oxygen species;
GSH glutathione; Dex, dexamethasone. |
Atorvastatin suppresses α-SMA
expression in lung tissues of asthmatic mice
To investigate the effect of atorvastatin treatment
on α-SMA expression in lung tissues of asthmatic mice, α-SMA
expression was measured by immunohistochemistry. As shown in
Fig. 8, OVA inhalation markedly
increased α-SMA expression in lung tissues of asthmatic mice, while
the administration of atorvastatin and Dex significantly decreased
the α-SMA expression in lung tissues. However, no significant
difference was observed between the atorvastatin group and the Dex
group (P>0.05).
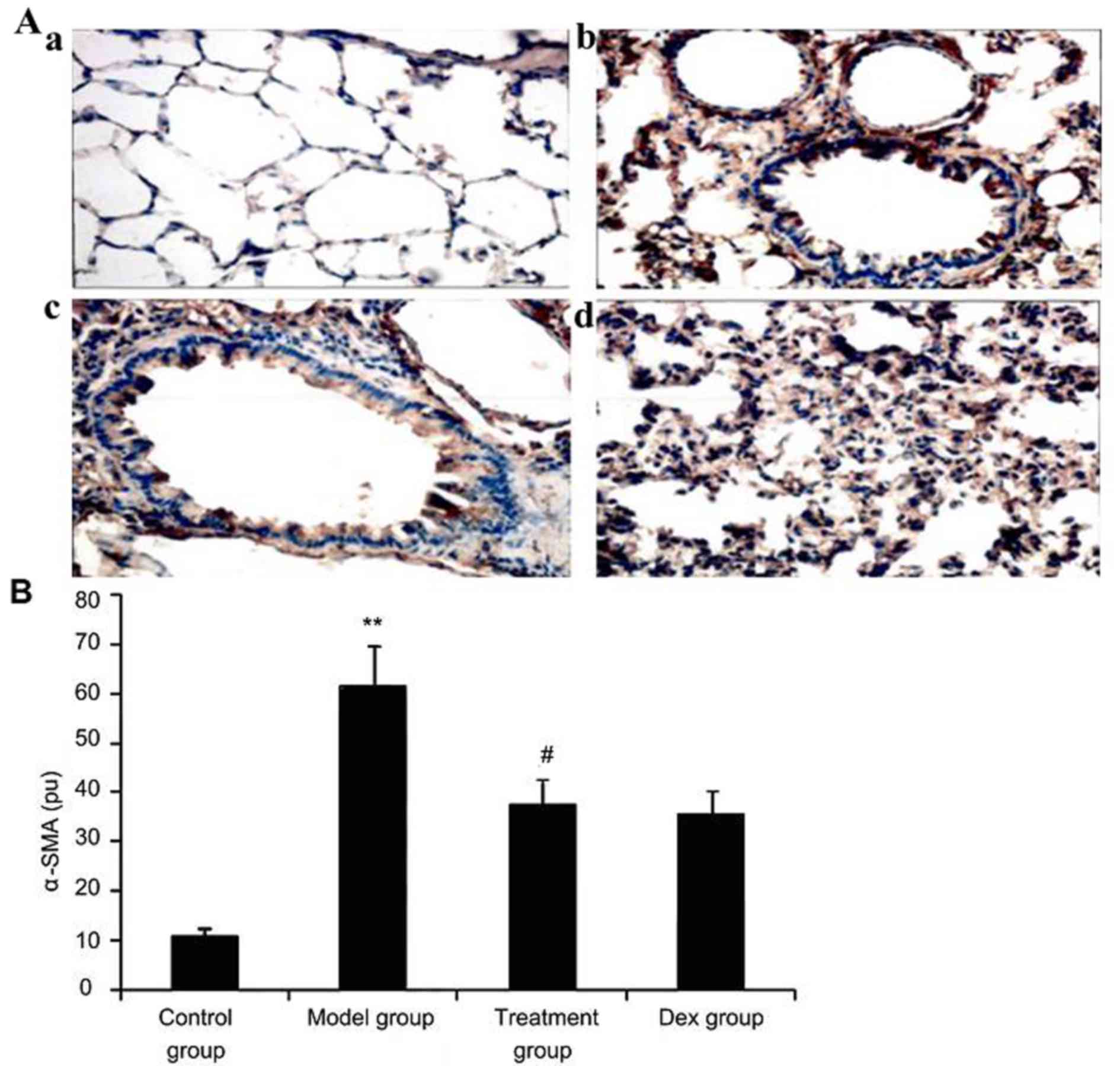 | Figure 8.Effect of atorvastatin treatment on
α-SMA expression in the lung tissues of asthmatic mice. Mice were
treated with atorvastatin or DEX and challenged with ovalbumin
(nebulized 2.5% solution 30 min/day, 3 days/week for 6 weeks), and
α-SMA expression in lung tissues was determined using
immunohistochemistry. (A) Representative immunostaining images
showing cells with positive expression levels of α-SMA in the four
groups of mice (immunofluorescence staining; magnification, ×400.
Groups: a, control group; b, model group; c, treatment group; d,
Dex group). (B) α-SMA-positive cells were quantified from the
immunohistochemical images to determine the expression of α-SMA in
lung tissues assayed. Values are expressed as the mean ± standard
deviation of one experiment consisting of three replicates. The
experiments were performed in triplicate. **P<0.05, vs. control
group; #P<0.05 vs. model group. Dex, dexamethasone;
SMA, smooth muscle actin; pu, positive units. |
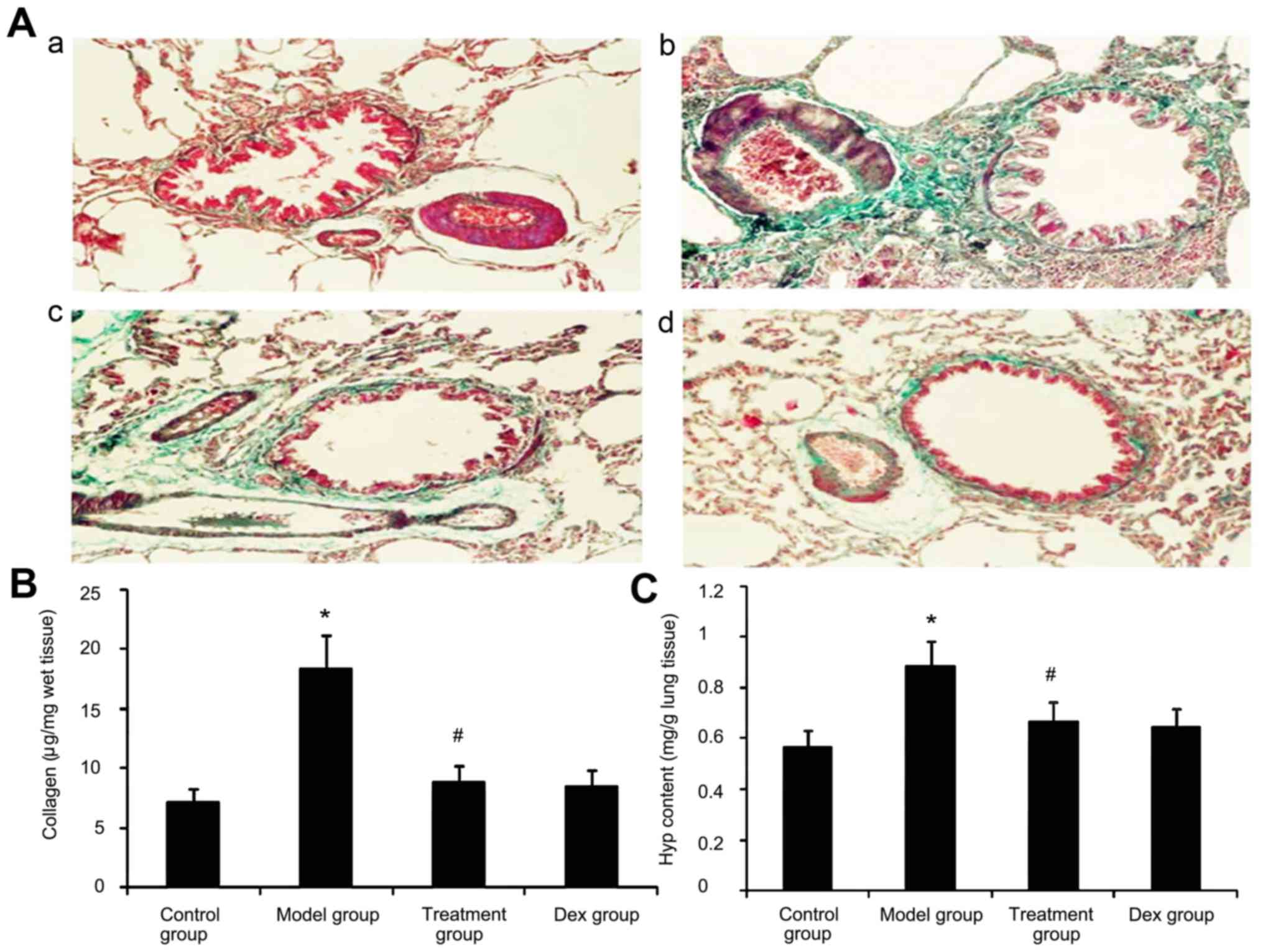
Effect of atorvastatin on lung
collagen deposition and Hyp content in asthmatic mouse lungs
Due to prolonged inflammation in chronic asthma,
increased collagen deposition is a hallmark of airway remodeling.
To evaluate the degree of collagen deposition, lung sections from
mice were stained with Masson's trichrome (Fig. 9A). In addition the Hyp content was
measured. As shown in Fig. 9, the
accumulation of collagen and Hyp content in lung tissues was
markedly increased in the OVA-challenged mice compared with that in
PBS-challenged mice. Treatment with Dex or atorvastatin
significantly reduced this increase (P<0.05). No significant
difference was observed between the atorvastatin group and the Dex
group (P>0.05).
Atorvastatin reduces the airway wall
area and ASM thickness
The results of the present study suggested that ASM
thickness may be associated with the severity of asthma. Therefore,
the airway wall area and ASM thickness in treated animals were
measured. As shown in Fig. 10,
OVA-challenged mice showed a significantly increased airway wall
area and ASM thickness compared with that in the control group
(P<0.05) and the treatment with Dex or atorvastatin
significantly reduced this increase (P<0.05). No significant
difference was observed between the atorvastatin and the Dex groups
(P>0.05). The airway wall area and ASM thickness in mice treated
with atorvastatin or Dex without OVA sensitization showed no
significant difference compared with the control group.
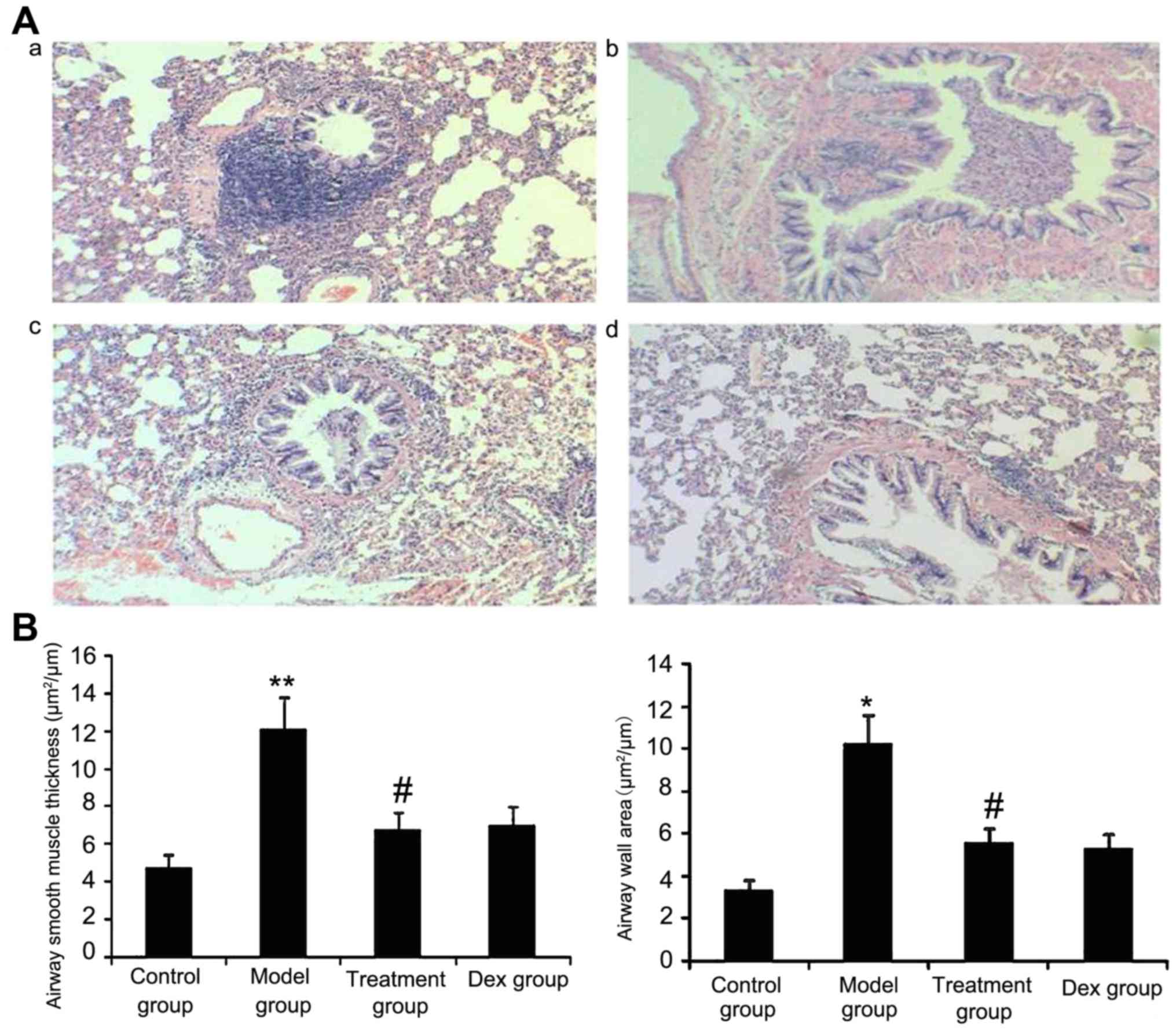 | Figure 10.Atorvastatin decreases the airway
wall area and airway smooth muscle thickness in mice. The mice were
treated with atorvastatin or Dex and challenged with ovalbumin
(nebulized 2.5% solution 30 min/day, 3 days/week for 6 weeks). For
the purpose of assessing the airway wall area and airway smooth
muscle thickness, tissue sections of the lungs of the mice were
stained with H&E (magnification, ×400). (A) Representative
images of the H&E-stained lung sections from the four
experimental groups are shown: a, control group; b, model group; c,
treatment group; d, Dex group. (B) Airway smooth muscle thickness
and airway wall area were determined using Image Pro-Plus software
6.0. Values are expressed as the mean ± standard deviation of one
experiment consisting of three replicates. The experiments were
performed in triplicate. *P<0.05, **P<0.01 vs. control group;
#P<0.05 vs. model group. Dex, dexamethasone; H&E,
hematoxylin and eosin. |
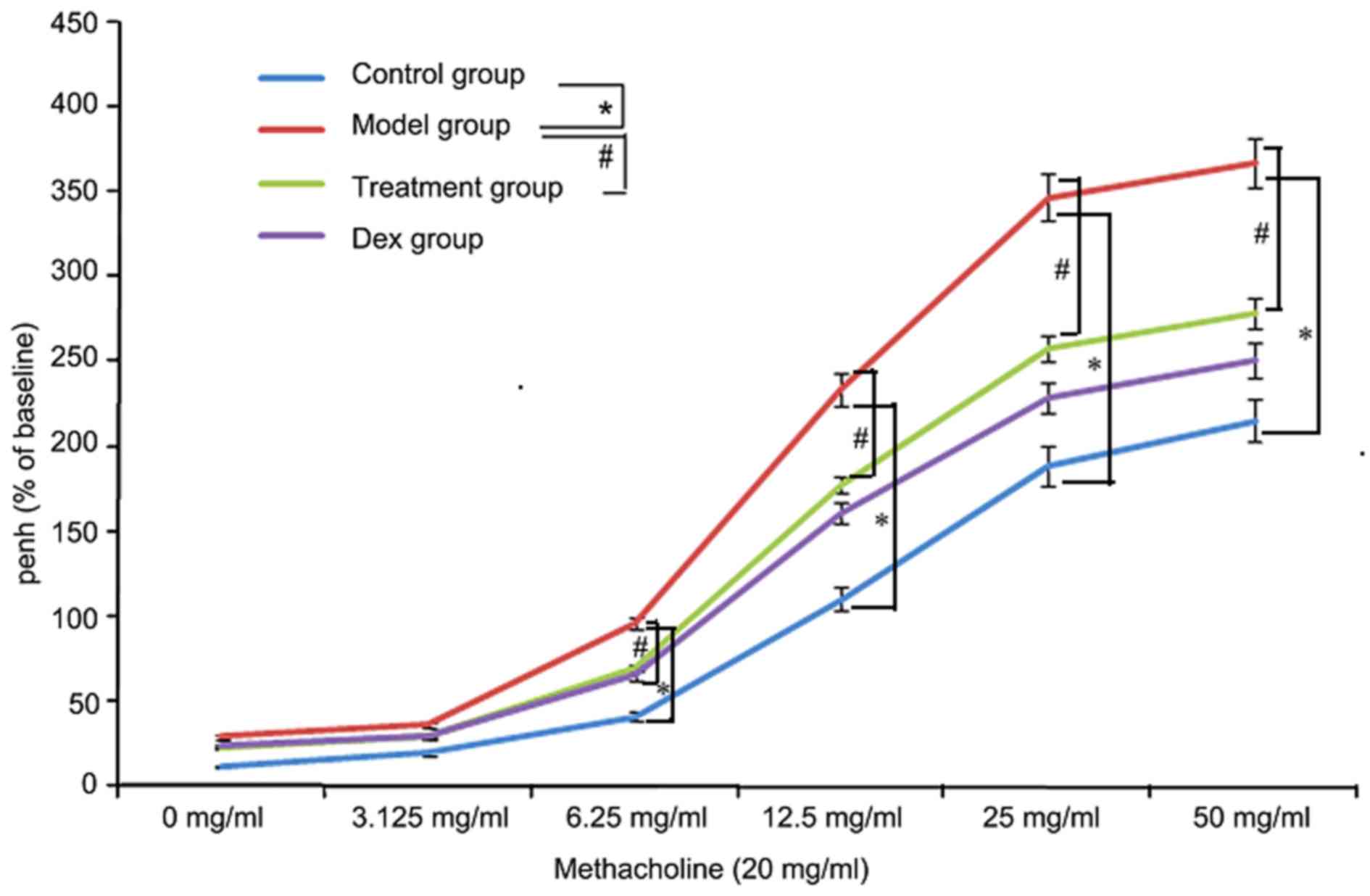
Atorvastatin reduces AHR
To investigate the effects of atorvastatin on AHR,
the physiological effect of the MCh administration on AHR was
examined. As shown in Fig. 11, the
Penh value of OVA-challenged mice was increased, as assessed by MCh
administration (20 mg/ml). However, the increased Penh response to
MCh challenge in OVA-challenged mice was effectively reduced by
atorvastatin administration.
Discussion
In the present study, a range of asthma-associated
symptoms was observed in the OVA-induced asthma mouse model. These
symptoms included altered levels of endurance, irritability,
shortness of breath, lips cyanosis, mouth breathing and astasia.
Pathologic observations indicated that the trachea and a large
number of inflammatory cells were present around alveoli and blood
vessels. In the acetylcholine provocation test, with increased
acetylcholine concentrations, the expiratory peak in the asthma
group is significantly decreased (27). This indicates that the airway
resistance is increased with elevated acetylcholine concentrations,
indicating the presence of bronchial hyperresponsiveness (BHR) in
the model. Lung tissue pathological biopsy showed bronchial wall
thickening and tracheal mucous membrane damage. Consequently, the
model used in the present study complies with the evaluation
criteria of successful bronchial asthma airway remodeling (17).
Bronchial remodeling is closely associated with
tracheal inflammation as a result of chronic airway inflammation
(28). This is caused by excessive
repair of chronic repetitive injuries to airway walls and is
characterized by variation in epithelial cell morphology, an
increase in the size of submucosal glands, hyperplasia and
hypertrophy of ASM, and hyperplasia of muscle fibroblasts (29). In the present study, the number of
pulmonary inflammatory cells, the generation of pro-inflammatory
mediators (IL-8, IL-17 and IL-13), and the expression levels of
intrapulmonary MMP-9 and TGF-β1 were all elevated. Moreover,
significant collagen deposition on the intrapulmonary bronchial
wall, bronchial remodeling and bronchial wall thickening led to
BHR.
Glucocorticoids are currently the optimal option to
control asthmatic inflammation (30). They alleviate trachea inflammation as
well as the release of inflammatory mediators (31), and inhibit or reverse airway
remodeling directly or indirectly through alleviating the
inflammation (32). Consequently, in
the present study, Dex treatment was applied in the positive
control group. The results of the present study showed that Dex
significantly inhibited trachea inflammation, reduced collagen
deposition on the bronchial wall, alleviated bronchial wall
thickening and improved bronchial remodeling in mice with bronchial
asthma.
Atorvastatin is a HMG-CoA reductase inhibitor
(33). The drug is the most widely
used and effective lipid-lowering drug. In addition to the
lipid-lowering effect, statins also has anti-inflammatory,
immunomodulatory and anti-BHR activities and inhibits cell
proliferation (33–37). The above effects are reflected in the
following aspects: Simvastatin can decrease the amount of BALF
cells in asthmatic mice and also reduces the levels of IL-4, IL-13
and TNF-α and the serum immunoglobulin E levels (33). Moreover, its anti-inflammatory
effects promote decreased airway resistance, improve lung
compliance and inhibit the generation and release of T-helper cell
type 1 (Th1) and Th2 factors (35,36).
However, the specific mechanism remains elusive. In the model used
in the present study, atorvastatin reduced the aggregation of
pulmonary inflammatory cells, decreased intrapulmonary
inflammation, increased the activity of antioxidant enzymes,
decreased the intrapulmonary oxidative stress response and reduced
the expression of genes involved in the stimulation of
intrapulmonary collagen deposition, thus improving bronchial
remodeling. Its effects were not significantly different from those
of Dex.
tTG participates in fibrotic processes in the lung,
kidney, liver and other visceral organs (6,38). TGF-β
is generally considered as one of the most critical cytokines
during fibrosis formation and development (39). It is a master regulator of fibrosis
formation and the strongest pro-fibrotic factor. tTG can cause
TGF-β1-linking protein to dissociate TGF-β1 from the ECM and
activate it (40). The activated
TGF-β1 can also increase tTG gene expression, forming a positive
feedback loop to increase the expression of collagen and
fibronectin, and promote the occurrence and development of fibrosis
(41). After pulmonary fibrosis was
induced by bleomycin in tTG knockout mice, the expression of MMP-9
decreased, thus reducing the generation of hydroxyproline and
collagen, and clearly relieving pulmonary fibrosis (41). In the present study, OVA
significantly promoted the increase of tTG levels in the bronchial
asthma mouse model, further increasing the activity of Smad3/TGF-β1
signaling, MMP-9 levels and collagen deposition to aggravate airway
remodeling. However, atorvastatin inhibited the expression of tTG,
reduced the activity of Smad3/TGF-β1 signaling and MMP-9 levels,
decreased collagen deposition and improved bronchial
remodeling.
The transcription factor NF-κB is at the core of the
inflammatory reaction (19). It
promotes the transcription and release of inflammatory factors
(19). Signal transduction occurs
from the interaction of TREM-1 with the SH2 structural domain of
the Syk tyrosine kinase. Syk phosphorylates CBL and growth factor
receptor-bound protein 2 to activate mitogen-activated protein
kinase, NF-κB and other transcription factors, promoting the
expression and secretion of inflammatory cytokines (12–42). In
the present study study, OVA was found to promote the expression of
TREM-1 of mice with bronchial asthma to further upregulate the
activity of NF-κB p65, accelerate the bronchial inflammatory
reaction of mice with bronchial asthma and aggravate bronchial wall
remodeling, and this was inhibited by atorvastatin.
The Nrf2 pathway is one of the most important
cellular resistance mechanisms in response to oxidative stress
damage in a variety of tissues and organs (43). NQO1 is a downstream antioxidant
enzyme in the Nrf2 pathway (44).
When a cell is subjected to oxidative stress, Nrf2 interacts with
kelch-like ECH-associated protein 1 to perform uncoupling prior to
being activated and transported into the cell nucleus. Nrf2 then
binds to antioxidant response elements in gene promoters to
regulate the expression of the downstream antioxidant enzyme NQO1,
thus enhancing the tolerance of cells to oxidative stress (45,46). The
pathogenesis of asthma is known to involve ROS generation and loss
of antioxidant defenses (43). A
widely recognized central feature of asthma and other airway
inflammatory diseases are alterations in alveolar and lung GSH
metabolism (45). In the present
study, atorvastatin increased the expression of Nrf2, NQO1 and
antioxidant enzyme GSH, reduced the expression of ROS and improved
bronchial remodeling in a mouse model.
MMP-9 promotes the synthesis of collagen IV and V as
well as laminin and elastin to form basement membrane ECM, and the
release of a variety of growth and differentiation factors (such as
TNF-α, VEGF and TGF-β), thus promoting tissue fibrosis, vascular
cell proliferation and tissue remodeling (47–49).
TIMP-1 is a specific inhibitor of MMP-9 and blocks substrate
degradation (48). In the present
study, atorvastatin inhibited the activity of MMP-9, reduced the
expression of VEGF and TGF-β, decreased collagen deposition in
bronchial airways in a mouse model of bronchial asthma and improved
bronchial remodeling in asthmatic mice. However, atorvastatin had a
lower impact on TIMP-1 levels than on the other mediators
examined.
TGF-β1 signaling has been identified as one of the
important mechanisms in bronchial remodeling (50). TGF-β1 expression is positively
correlated to the number of fibroblasts and bronchial basement
membrane thickness, as well as asthma severity. Smad3 promotes the
expression of TGF-β1 and bronchial remodeling (51). In the present study, atorvastatin
inhibited the phosphorylation of Smad3 and reduced the expression
of TGF-β1.
VEGF participates in inflammatory cell movement,
maintains cell survival and promotes the expression of TGF-β1
(52). The expression of VEGF under
low-oxygen conditions is regulated by HIF-lα (53). In the present study, the expression
of HIF-1α in the lungs of bronchial asthmatic mice increased and is
the likely reason for the elevated expression of VEGF and TGF-β1 as
well as the aggravation of bronchial remodeling in mice with
bronchial asthma. Furthermore, atorvastatin treatment blocked the
expression of HIF-1α, and reduced the expression of VEGF and
TGF-β1.
α-SMA is the predominant isoform of actin within
vascular smooth-muscle cells and has an important role in bronchial
remodeling; it is also a major marker of ASM, with reduced levels
reflecting changes of ASM quantity and shrinking (54). The present study showed that
atorvastatin decreased α-SMA levels in the mouse model of bronchial
asthma.
Hyp is a major component of the protein collagen
(55). Hyp and proline have key
roles in collagen stability (54).
The increase in bronchial wall thickness in asthma is associated
with the deposition of collagen and other matrix proteins, such as
fibronectin, cytotactin, tendon protein and laminin. Airway wall
thickening is correlated with clinically severe asthma (55) and is a prominent feature of lung
tissue of patients with fatal asthma. In the present study, the Hyp
content and deposition of collagen in asthmatic mouse lungs was
increased, which was suppressed by atorvastatin.
AHR is positively correlated with the severity of
bronchial remodeling and asthma (53). A reduction in AHR is an important
indicator of therapeutic benefit. The results of the present study
indicated that atorvastatin decreased AHR by improving bronchial
remodeling. Bronchial resistance in the atorvastatin group was
obviously lower than that in the OVA group, indicating that
atorvastatin reduces the BHR of asthmatic mice.
In conclusion, the present study demonstrated that
atorvastatin inhibited asthma-associated airway wall remodeling
through mechanisms involving a decrease in the expression of tTG
and TREM-1 as well as modulation of the Nrf2 signaling pathway in
the lung. This suggests the possibility of further developing
atorvastatin as a candidate for the prevention and treatment of
airway remodeling in asthma.
Glossary
Abbreviations
Abbreviations:
|
TREM-1
|
triggering receptor expressed on
myeloid cells 1
|
|
BHR
|
bronchial hyperresponsiveness
|
|
tTG
|
tissue transglutaminase
|
|
VEGF
|
vascular endothelial growth factor
|
|
NF-κB
|
nuclear factor kappa B
|
|
MMP-9
|
matrix metalloproteinase 9
|
|
DEX
|
dexamethasone
|
|
α-SMA
|
smooth muscle actin-α
|
|
NQO1
|
NADPH quinine oxidoreductase
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
HIF-1α
|
hypoxia-inducible factor 1α
|
|
TGF-β
|
transforming growth factor β
|
|
BALF
|
bronchoalveolar lavage fluid
|
References
|
1
|
Befekadu E, Onofrei C and Colice GL:
Tiotropium in asthma: A systematic review. J Asthma Allergy.
7:11–21. 2014.PubMed/NCBI
|
|
2
|
Roche WR, Beasley R, Williams JH and
Holgate ST: Subepithelial fibrosis in the bronchi of asthmatics.
Lancet. 1:520–524. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aikawa T, Shimura S, Sasaki H, Ebina M and
Takishima T: Marked goblet cell hyperplasia with mucus accumulation
in the airways of patients who died of severe acute asthma attack.
Chest. 101:916–921. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wiggs BR, Bosken C, Paré PD, James A and
Hogg JC: A model of airway narrowing in asthma and in chronic
obstructive pulmonary disease. Am Rev Respir Dis. 145:1251–1258.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nakaoka H, Perez DM, Baek KJ, Das T,
Husain A, Misono K, Im MJ and Graham RM: Gh: A GTP-binding protein
with transglutaminase activity and receptor signaling function.
Science. 264:1593–1596. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lorand L and Graham RM: Transglutaminases:
Crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell
Biol. 4:140–156. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iismaa SE, Mearns BM, Lorand L and Graham
RM: Transglutaminases and disease: Lessons from genetically
engineered mouse models and inherited disorders. Physiol Rev.
89:991–1023. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Griffin M, Casadio R and Bergamini CM:
Transglutaminases: Nature's biological glues. Biochem J.
368:377–396. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shweke N, Boulos N, Jouanneau C,
Vandermeersch S, Melino G, Dussaule JC, Chatziantoniou C, Ronco P
and Boffa JJ: Tissue transglutaminase contributes to interstitial
renal fibrosis by favoring accumulation of fibrillar collagen
through TGF-beta activation and cell infiltration. Am J Pathol.
173:631–642. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Radsak MP, Salih HR, Rammensee HG and
Schild H: Triggering receptor expressed on myeloid cells-1 in
neutrophil inflammatory responses: Differential regulation of
activation and survival. J Immunol. 172:4956–4963. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bouchon A, Dietrich J and Colonna M:
Cutting edge: Inflammatory responses can be triggered by TREM-1, a
novel receptor expressed on neutrophils and monocytes. J Immunol.
164:4991–4995. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bouchon A, Facchetti F, Weigand MA and
Colonna M: TREM-1 amplifies inflammation and is a crucial mediator
of septic shock. Nature. 410:1103–1107. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fassett RG, Robertson IK, Ball MJ,
Geraghty DP and Coombes JS: Effects of atorvastatin on biomarkers
of inflammation in chronic kidney disease. Clin Nephrol. 81:75–85.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Simpson RJ, Signorovitch J, Ramakrishnan
K, Ivanova J, Birnbaum H and Kuznik A: Cardiovascular and economic
outcomes after initiation of atorvastatin versus simvastatin in an
employed population stratified by cardiovascular risk. Am J Ther.
18:436–448. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Balli U, Keles GC, Cetinkaya BO, Mercan U,
Ayas B and Erdogan D: Assessment of vascular endothelial growth
factor and matrix metalloproteinase-9 in the periodontium of rats
treated with atorvastatin. J Periodontol. 85:178–187. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Feldman C: Statins for non-cystic fibrosis
bronchiectasis. Lancet Respir Med. 2:431–432. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jain W, Kitagaki K, Businga T, Hussain I,
George C, O'shaughnessy P and Kline JN: CpG-oligodeoxynucleotides
inhibit airway remodeling in a murine model of chronic asthma. J
Allergy Clin Immunol. 110:867–872. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu MW, Wang YH, Qian CY and Li H:
Xuebijing exerts protective effects on lung permeability leakage
and lung injury by upregulating Toll-interacting protein expression
in rats with sepsis. Int J Mol Med. 34:1492–1504. 2014.PubMed/NCBI
|
|
20
|
Mizutani N, Nabe T and Yoshino S: IL-17A
promotes the exacerbation of il-33-induced airway
hyperresponsiveness by enhancing neutrophilic inflammation via
CXCR2 signaling in mice. J Immunol. 192:1372–1384. 2004. View Article : Google Scholar
|
|
21
|
Entezari M, Javdan M, Antoine DJ, Morrow
DM, Sitapara RA, Patel V, Wang M, Sharma L, Gorasiya S, Zur M, et
al: Inhibition of extracellular HMGB1 attenuates hyperoxia-induced
inflammatory acute lung injury. Redox Biol. 2:314–322. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wen H, Gwathmey JK and Xie LH: Oxidative
stress-mediated effects of angiotensin II in the cardiovascular
system. World J Hypertens. 2:34–44. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wei B, Shang YX, Li M, Jiang J and Zhang
H: Cytoskeleton changes of airway smooth muscle cells in juvenile
rats with airway remodeling in asthma and the RhoA/ROCK signaling
pathway mechanism. Genet Mol Res. 13:559–569. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jamall IS, Finelli VN and Que Hee SS: A
simple method to determine nanogram levels of 4-hydroyproline in
biological tissues. Anal Biochem. 112:70–75. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bai A, Eidelman DH, Hogg JC, James AL,
Lambert RK, Ludwig MS, Martin J, McDonald DM, Mitzner WA, Okazawa
M, et al: Proposed nomenclature for quantifying subdivisions of the
bronchial wall. J Appl Physiol (1985). 77:1011–1014.
1994.PubMed/NCBI
|
|
26
|
Takeda N, Kondo M, Ito S, Ito Y, Shimokata
K and Kume H: Role of RhoA inactivation in reduced cell
proliferation of human airway smooth muscle by simvastatin. Am J
Respir Cell Mol Biol. 35:722–729. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mookerjee I, Solly NR, Royce SG, Tregear
GW, Samuel CS and Tang ML: Endogenous relaxin regulates collagen
deposition in an animal model of allergic airway disease.
Endocrinology. 147:754–761. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Davies DE and Holgate ST: Asthma: The
importance of epithelial mesenchymal communication in pathogenesis.
Inflammation and the airway epithelium in asthma. Int J Biochem
Cell Biol. 34:1520–1526. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang ML, Wilson JW, Stewart AG and Royce
SG: Airway remodelling in asthma: Current understanding and
implications for future therapies. Pharmacol Ther. 112:474–488.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Barnes PJ: Anti-inflammatory actions of
glucocorticoids: Molecular mechanisms. Clin Sci (Lond). 94:557–572.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boulet LP, Turcotte H, Laviolette M, Naud
F, Bernier MC, Martel S and Chakir J: Airway hyperresponsiveness,
inflammation, and subepithelial collagen deposition in recently
diagnosed versus long-standing mild asthma. Influence of inhaled
corticosteroids. Am J Respir Crit Care Med. 162:1308–1313. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Irwin RS and Richardson ND: Side effects
with inhaled corticosteroids: The physician's perception. Chest.
130 1 Suppl:41S–53S. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Johnson BA, Iacono AT, Zeevi A, McCurry KR
and Duncan SR: Statin use is associated with improved function and
survival of lung allografts. Am J Respir Crit Care Med.
167:1271–1278. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McKay A, Leung BP, McInnes IB, Thomson NC
and Liew FY: A novel anti-inflammatory role of simvastatin in a
murine model of allergic asthma. J Immunol. 172:2903–2908. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zeki AA, Franzi L, Last J and Kenyon NJ:
Simvastatin inhibits airway hyperreactivity: Implications for the
mevalonate pathway and beyond. Am J Respir Crit Care Med.
180:731–740. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chiba Y, Arima J, Sakai H and Misawa M:
Lovastatin inhibits bronchial hyperresponsiveness by reducing RhoA
signaling in rat allergic asthma. Am J Physiol Lung Cell Mol
Physiol. 294:L705–L713. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chiba Y, Sato S and Misawa M: Inhibition
of antigen-induced bronchial smooth muscle hyperresponsiveness by
lovastatin in mice. J Smooth Muscle Res. 44:123–128. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sánchez-Lara AC, Elliott J, Syme HM, Brown
CA and Haylor JL: Feline chronic kidney disease is associated with
upregulation of transglutaminase 2: A collagen cross-linking
enzyme. Vet Pathol. 52:513–523. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sime PJ and O'Reilly KM: Fibrosis of the
lung and other tissues: New concepts in pathogenesis and treatment.
Clin Immunol. 99:308–319. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Griffin M, Smith LL and Wynne J: Changes
in transglutaminase activity in an experimental model of pulmonary
fibrosis induced by paraquat. Br J Exp Pathol. 60:653–661.
1979.PubMed/NCBI
|
|
41
|
Olsen KC, Sapinoro RE, Kottmann RM,
Kulkarni AA, Iismaa SE, Johnson GV, Thatcher TH, Phipps RP and Sime
PJ: Transglutaminase 2 and its role in pulmonary fibrosis. Am J
Respir Crit Care Med. 184:699–707. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Genua M, Rutella S, Correale C and Danese
S: The triggering receptor expressed on myeloid cells (TREM) in
inflammatory bowel disease pathogenesis. J Transl Med. 12:2932014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Erlank H, Elmann A, Kohen R and Kanner J:
Polyphenols activate Nrf2 in astrocytes via H2O2, semiquinones, and
quinones. Free Radic Biol Med. 51:2319–2327. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Park JS, Jung JS, Jeong YH, Hyun JW, Le
TK, Kim DH, Choi EC and Kim HS: Antioxidant mechanism of isoflavone
metabolites in hydrogen peroxide-stimulated rat primary astrocytes:
Critical role of hemeoxygenase-1 and NQO1 expression. J Neurochem.
119:909–919. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hou DX, Korenori Y, Tanigawa S,
Yamada-Kato T, Nagai M, He X and He J: Dynamics of Nrf2 and Keap1
in ARE-mediated NQO1 expression by wasabi 6-(methylsulfinyl)hexyl
isothiocyanate. J Agric Food Chem. 59:11975–11982. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee IS, Lim J, Gal J, Kang JC, Kim HJ,
Kang BY and Choi HJ: Anti-inflammatory activity of xanthohumol
involves heme oxygenase-1 induction via NRF2-ARE signaling in
microglial BV2 cells. Neurochem Int. 58:153–160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Barbaro MP, Spanevello A, Palladino GP,
Salerno FG, Lacedonia D and Carpagnano GE: Exhaled matrix
metalloproteinase-9 (MMP-9) in different biological phenotypes of
asthma. Eur J Intern Med. 25:92–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Possa SS, Charafeddine HT, Righetti RF, da
Silva PA, Almeida-Reis R, Saraiva-Romanholo BM, Perini A, Prado CM,
Leick-Maldonado EA, Martins MA and Tibério Ide F: Rho-kinase
inhibition attenuates airway responsiveness, inflammation, matrix
remodeling, and oxidative stress activation induced by chronic
inflammation. Am J Physiol Lung Cell Mol Physiol. 303:L939–L952.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yeo NK, Eom DW, Oh MY, Lim HW and Song YJ:
Expression of matrix metalloproteinase 2 and 9 and tissue inhibitor
of metalloproteinase 1 in nonrecurrent vs recurrent nasal polyps.
Ann Allergy Asthma Immunol. 111:205–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Che Z, Zhu X, Yao C, Liu Y, Chen Y, Cao J,
Liang C and Lu Y: The association between the C-509T and T869C
polymorphisms of TGF-β1 gene and the risk of asthma: A
meta-analysis. Hum Immunol. 75:141–150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Baek KJ, Cho JY, Rosenthal P, Alexander
LE, Nizet V and Broide DH: Hypoxia potentiates allergen induction
of HIF-1α, chemokines, airway inflammation, TGF-β1, and airway
remodeling in a mouse model. Clin Immunol. 147:27–37. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mirzoeva S, Franzen CA and Pelling JC:
Apigenin inhibits TGF-β-induced VEGF expression in human prostate
carcinoma cells via a Smad2/3- and Src-dependent mechanism. Mol
Carcinog. 53:598–609. 2014.PubMed/NCBI
|
|
53
|
Le Cras TD, Acciani TH, Mushaben EM,
Kramer EL, Pastura PA, Hardie WD, Korfhagen TR, Sivaprasad U,
Ericksen M, Gibson AM, et al: Epithelial EGF receptor signaling
mediates airway hyperreactivity and remodeling in a mouse model of
chronic asthma. Am J Physiol Lung Cell Mol Physiol. 300:L414–L421.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Park SJ, Lee KS, Lee SJ, Kim SR, Park SY,
Jeon MS, Lee HB and Lee YC: L-2-Oxothiazolidine-4-carboxylic acid
or α-lipoic acid attenuates airway remodeling: Involvement of
nuclear factor-κB (NF-κB), nuclear factor erythroid 2p45-related
factor-2 (Nrf2), and hypoxia-inducible factor (HIF). Int J Mol Sci.
13:7915–7937. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fuchimoto Y, Kanehiro A, Miyahara N, Koga
H, Ikeda G, Waseda K, Tanimoto Y, Ueha S, Kataoka M, Gelfand EW and
Tanimoto M: Requirement for chemokine receptor 5 in the development
of allergen-induced airway hyperresponsiveness and inflammation. Am
J Respir Cell Mol Biol. 45:1248–1255. 2011. View Article : Google Scholar : PubMed/NCBI
|