Introduction
The mammalian target of rapamycin (mTOR) has
attracted attention due to its critical role in cellular life
sustaining activity and vital involvement in a variety of diseases
(1). Deregulation of the mTOR
signaling pathway affects essential cellular processes and is
associated with a number of pathological conditions, including
cancer, obesity, type 2 diabetes, and neuron diseases (2). In the neuron system, decreased mTOR
signaling is associated with neurodegeneration (3), whereas excess activation of mTOR
signaling leads to the abnormal development of neurons and glia,
which results in brain malformation (4).
Although mTOR provides positive effects on anabolic
processes, growth may be stimulated by negatively regulating
autophagy, the central degradative process in cells (2). Autophagy is involved in recycling
damaged organelles and is required for the organismal and cellular
adaptation to nutrient starvation (2,5). This
conserved process aids the maintenance of cellular homeostasis by
removing damaged or toxic intrinsic components (6). Furthermore, several studies have
suggested that intracellular protein degradation pathways, such as
autophagy, are deregulated in neurodegenerative diseases and may
possess vital roles in the etiology of neuron-related pathologies
(7–9). mTOR signaling is considered to be one
of the major pathways involved in the regulation of autophagy and
the implications of mTOR signaling in neurodegenerative diseases
has been investigated over the last decade (10). Moreover, mTOR negatively regulates
the biogenesis of lysosomes, a group of multifunctional organelles
that have the capacity to degrade many types of cellular components
(11–13).
Rapamycin is a specific inhibitor of mTOR that acts
by binding to the FK506-binding protein 12 kDa (FKBP12), forming a
molecular complex that impairs mTOR activity (14,15).
mTOR kinase inhibitors such as rapamycin are more efficient than
the first generation of rapalogs in promoting autophagy and
blocking protein synthesis (16),
and it has therefore been suggested that mTOR kinase inhibitors may
be more effective in treating diseases associated with the
formation and accumulation of protein aggregates. Furthermore,
rapamycin is able to reduce the rate of protein synthesis and thus
hinder the aggregation of misfolded proteins. This implies that
alternative effectors of mTOR signaling may have roles in the
development of neuron-related pathologies (17).
Additionally, the promotion of autophagy linked to
mTOR inhibition could mediate some effects of mTOR on longevity. A
previous study has indicated that suppression of autophagy gene
activity hindered the life span extension in worms with aberrant
TOR signaling (18). Therefore,
inducing autophagy may reduce the aging process while promoting the
breakdown of abnormal proteins and damaged organelles. In a
previous report, the Interventions Testing Program (ITP) from the
National Institute on Aging demonstrated that rapamycin-induced
inhibition of mTOR expanded the life span in mice (19,20).
Therefore, it was hypothesized that the mTOR
signaling pathway may possess a key role in the neurodegenerative
and aging process of senescence accelerated mouse-prone 8 (SAMP8)
mice. Furthermore, treatment with rapamycin and regulating the mTOR
pathway may improve the neurodegenerative changes. To test this
hypothesis, phosphorylation levels of mTOR and its substrate, p70S6
kinase (p70S6K) was detected. Additionally, the expression levels
of anti-apoptotic protein, B cell lymphoma-2 (Bcl-2), were
evaluated to investigate the alteration of mTOR signaling pathway
in aged SAMP8 mice and primary neurons. As the approach of inducing
autophagy has been suggested to aid the removal of abnormal protein
aggregates and promote survival in neurons, we investigated whether
autophagy may be neuroprotective via preventing cell death in SAMP8
mice.
Materials and methods
Antibodies and reagent
Mouse monoclonal antibodies (mAbs) for Tau (Tau46)
(cat. no. 4019) and phospho-Tau (Ser396) (cat. no. 9632) and rabbit
mAbs for beclin-1 (D40C5) (cat. no. 3495), phospho-mTOR (Ser2448)
(cat. no. 2971), and phospho-p70 S6 (Thr389) (cat. no. 9205) were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Rabbit polyclonal antibody (pAb) phospho-Tau (Ser199) (cat. no.
44-734G) was obtained from Thermo Fisher Scientific, Inc. (Waltham,
MA, USA). Mouse mAb MAP 2a, b, c (Microtubule-Associated Protein)
Ab-3 (cat. no. MS-250-P) was from Thermo Fisher Scientific, Inc.
Rabbit pAb for LC3B (cat. no. ab48394) was supplied from Abcam
(Cambridge, MA, USA). Rabbit pAbs for mTOR (P2476) (cat. no.
BS1555) and Bcl-2 (cat. no. BS1511) were purchased from Bioworld
Technology, Inc. (St. Louis Park, MN, USA). Infrared-labeled second
antibodies (cat. no. 611-731-127) were obtained from LI-COR
Biosciences, Inc. (Lincoln, NE, USA). B-27 Serum-free supplement,
fetal bovine serum (FBS) and Neurobasal-A medium were supplied from
Thermo Fisher Scientific, Inc. CelLytic MT Cell Lysis Reagent was
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Animals
Newborn male SAMP8 and SAMR1 mice were purchased,
with six mice in each group. All the mice were maintained and
exposed to a 12-h light-dark cycle, had free access to the food and
water ad libitum and were kept at a temperature of 20–22°C.
The present study was conducted in accordance with the guidelines
established by the Local Animal Care and Use committee.
Primary neuron culture
Primary cultured neurons were established using
hippocampus tissues from one to three-day-old SAMP8 and SAMR1 mice.
Mice were then sacrificed by decapitation to avoid damaging the
brain, and fully immersed in 75% alcohol for disinfection. An
incision was made in the scalp and skull to expose brain and the
brain tissue was placed in ice-cold dissection solution. The
hippocampus was isolated and dissected free of the meninges and
blood vessels under a dissecting microscope. The tissue was diced
into small cubes and dissociated by incubation with 0.125% trypsin
for 15–25 min at 37°C. Dulbecco's modified Eagle's medium (DMEM),
supplemented with 10% FBS to terminate the digestion, was
subsequently added. The tissue was left to precipitate using
repeated blows with a polished glass pipette and the supernatant
was transferred to a new tube. The supernatant was centrifuged at
114.6 × g at 16°C for 5 min, discarded and the DMEM containing 10%
FBS was added to the precipitation to make a single cell
suspension. Cells were seeded in 35-mm dishes
(3.37×105/cm2) at 37°C in a humidified
atmosphere of 5% CO2. Overnight, the culture medium was
replaced with Neurobasal-A medium containing 2% B27 serum-free
medium. The culture medium was changed every three days.
At the seventh day of culturing the cells, neurons
from SAMP8 mice were treated with either 0.5 or 1.0 µM rapamycin
(Sigma-Aldrich; Merck KGaA) and plated in culture medium for three
days. The neuron cells were fixed in phosphate-buffered saline
(PBS) followed by either immunofluorescence or western-blot
analysis. There were four groups in this part of experiment (n=6
per group): Neurons-SAMR1 group; neurons-SAMP8 group; neurons-SAMP8
+ 0.5 µM rapamycin group; and neurons-SAMP8 + 1.0 µM rapamycin
group.
Immunocytochemistry
At the tenth day, neurons cultured in dishes were
fixed for 30 min with 4% paraformaldehyde in PBS. Cells were washed
three times in PBS and made permeable with 0.5% Triton X-100 and 5%
goat serum (Invitrogen; Thermo Fisher Scientific, Inc.) in PBS for
40 min, at room temperature. Immunolabeling was performed with
mouse mAb MAP 2a, b, c Ab-3 (1:100), at 4°C overnight. Cells were
washed three times in PBS and incubated with TRITIC-labeled
secondary antibody (1:100; cat. no. ZF0313; Beijing Xinqiao
Technology Development Co., Ltd., Beijing, China) at 4°C for 10
days. Labeled cells were examined using an inverted fluorescence
microscope (Nikon Corporation, Tokyo, Japan) with an NIS imaging
analysis system (Nikon Corporation). Neurons were indicated by red
fluorescence.
Protein extraction
Animals were anesthetized with intraperitoneal
injection of 2% sodium pentobarbital (40–50 mg/kg), then sacrificed
by decapitation. The cerebral cortex and hippocampus were
dissected, immediately weighed, frozen in liquid nitrogen, and
stored at −80°C. Protein extracts were prepared in RIPA cell lysis
buffer (Thermo Fisher Scientific, Inc.). The lysates were sonicated
in an iced water bath for 30 min and centrifuged at 37,318.4 × g at
4°C for 20 min to collect the supernatants. The protein
concentration was measured using a BCA protein assay reagent
(Sigma-Aldrich; Merck KGaA) according to manufacturer's
instructions.
Western blot analysis
Protein extracts (100 µg/lane) were separated using
10% SDS-PAGE, transferred to polyvinylidene difluoride (PVDF)
membranes. Membranes were incubated with 5% non-fat milk and TBS
(0.1%) Tween-20 blocking solution for 2 h at room temperature
overnight and then washed with TBS (0.1%) Tween-20 blocking
solution for 30 min at room temperature with gentle shaking.
Membranes were probed with primary antibodies against LC3B (1:500),
beclin 1 (1:500), TAU (pS396) (1:500), TAU (pS199) (1:500), Tau46
(1:1,000), Phospho-mTOR (Ser2448) (1:1,000), mTOR (1:500), p70S6K
(pThr389) (1:500), Bcl-2 (1:500) or β-actin (1:300). All primary
antibodies were diluted with blocking solution and incubated
overnight at 4°C. After washing with TBS-0.1% Tween-20, the PVDF
membranes were incubated with fluorescently-labeled secondary
antibodies (1:10,000, diluted in blocking solution) for 1 h at room
temperature, and signals were detected with an Odyssey Infrared
Imaging System (LI-COR Biosciences, Inc.). Band quantitation of
western blots was determined using the image analysis system,
Quantity One (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
β-actin protein was used as control. Each experiment was repeated
three times with samples from three different mice.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical significance was evaluated by a one-way analysis of
variance or via the Non-parametric test. Analyses were conducted
using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
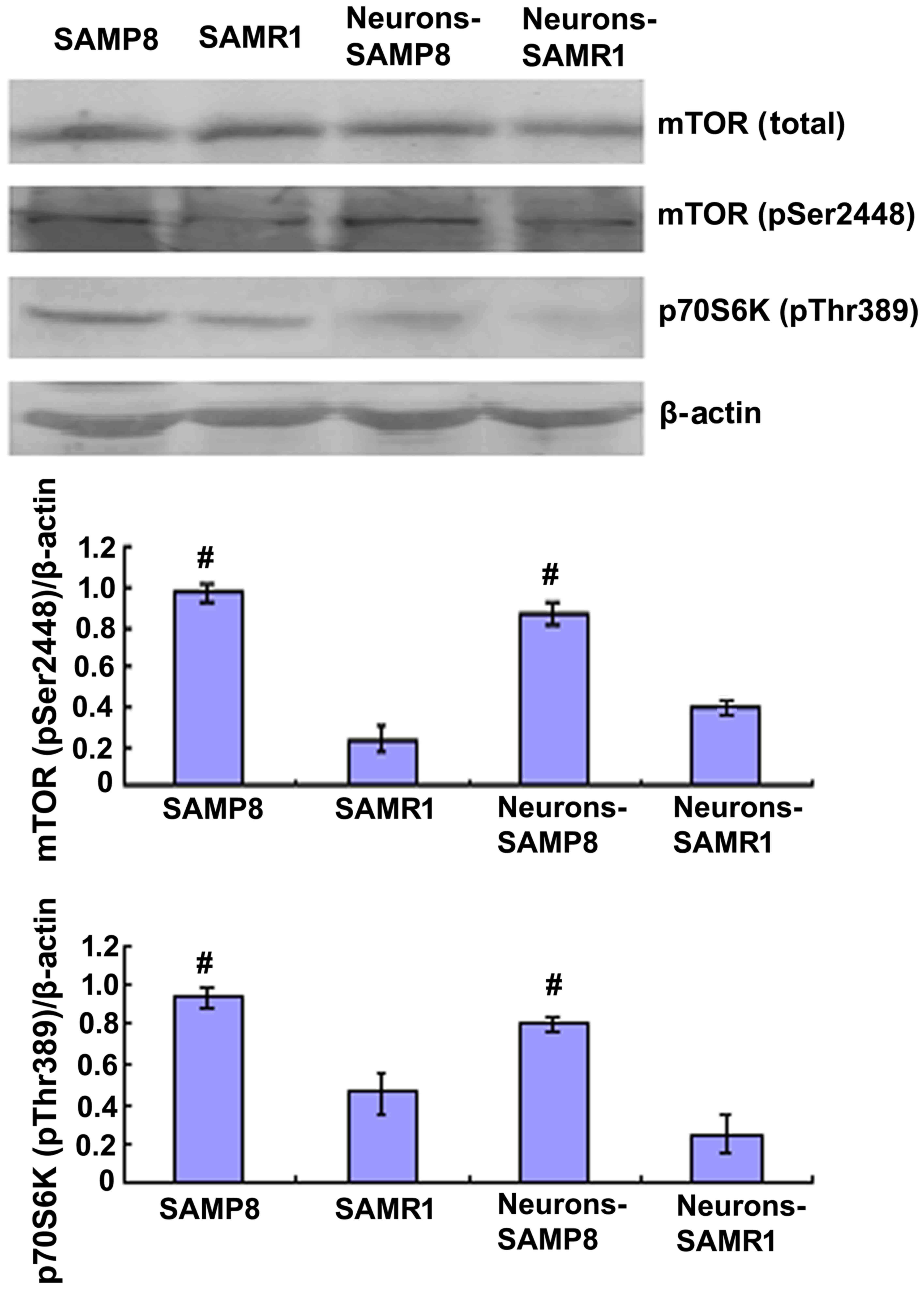
Upregulation of mTOR signaling in aged
SAMP8 compared with SAMR1
In order to determine the changes in mTOR signaling
in the aging of SAMP8 mice, western blot analysis was performed to
analyze the protein expression levels of total mTOR and mTOR
phosphorylation at Ser2448. Western blotting data indicated that
the protein expression levels of mTOR (pSer2448) in the SAMP8 group
were significantly increased when compared with the control SAMR1
group in vitro (P<0.01; Fig.
1).
The 70-kDa S6 kinase (p70S6K) is a Ser/Thr-directed
kinase that has a vital role in cell growth, differentiation, and
cycle control (21). mTOR function
is typically established via the steady-state level of
phosphorylated p70S6K at Thr389, an epitope which is specifically
phosphorylated by mTOR; however, mTOR phosphorylation does not
consistently rely on this activity (22,23).
Western blot analysis revealed that the protein expression levels
of phosphorylated p70S6K in the hippocampal primary neurons of
SAMP8 mice were significantly increased when compared with the
control SAMR1 group, indicating enhanced mTOR activity in aged mice
(Fig. 1).
Rapamycin treatment promoted cell
morphology and alleviated Tau phosphorylation of neurons from
SAMP8
To determine whether or not mTOR signaling had a
direct correlation with the neurodegenerative alterations in
primary cultured neurons of SAMP8 mice, immunofluorescence staining
was used to observe the cell morphology of neurons when cultured at
the tenth day or pretreated with rapamycin for three days. Neurons
derived from the control SAMR1 group appeared to be normal with
slender and smooth projections (Fig.
2Aa) whereas, neurons extracted from SAMP8 were in a poor
state, with fragmented or bead-like processes (Fig. 2Ab). When pretreated with 0.5 µM
rapamycin, a well-known mTOR inhibitor, the neurons from SAMP8
exhibited an improved appearance when compared with the non-treated
SAMP8 group, with smooth and slender projections in some of the
neurons (Fig. 2Ac). When pretreated
with 1.0 µM rapamycin, the neurons exhibited a poorer cell state
when compared with the untreated SAMP8 group, with most neurons
lacking projections (Fig. 2Ad). The
present findings suggested that 1.0 µM rapamycin may be harmful to
neurons, therefore 0.5 µM rapamycin was used to pretreat the
neurons from SAMP8 in subsequent experiments.
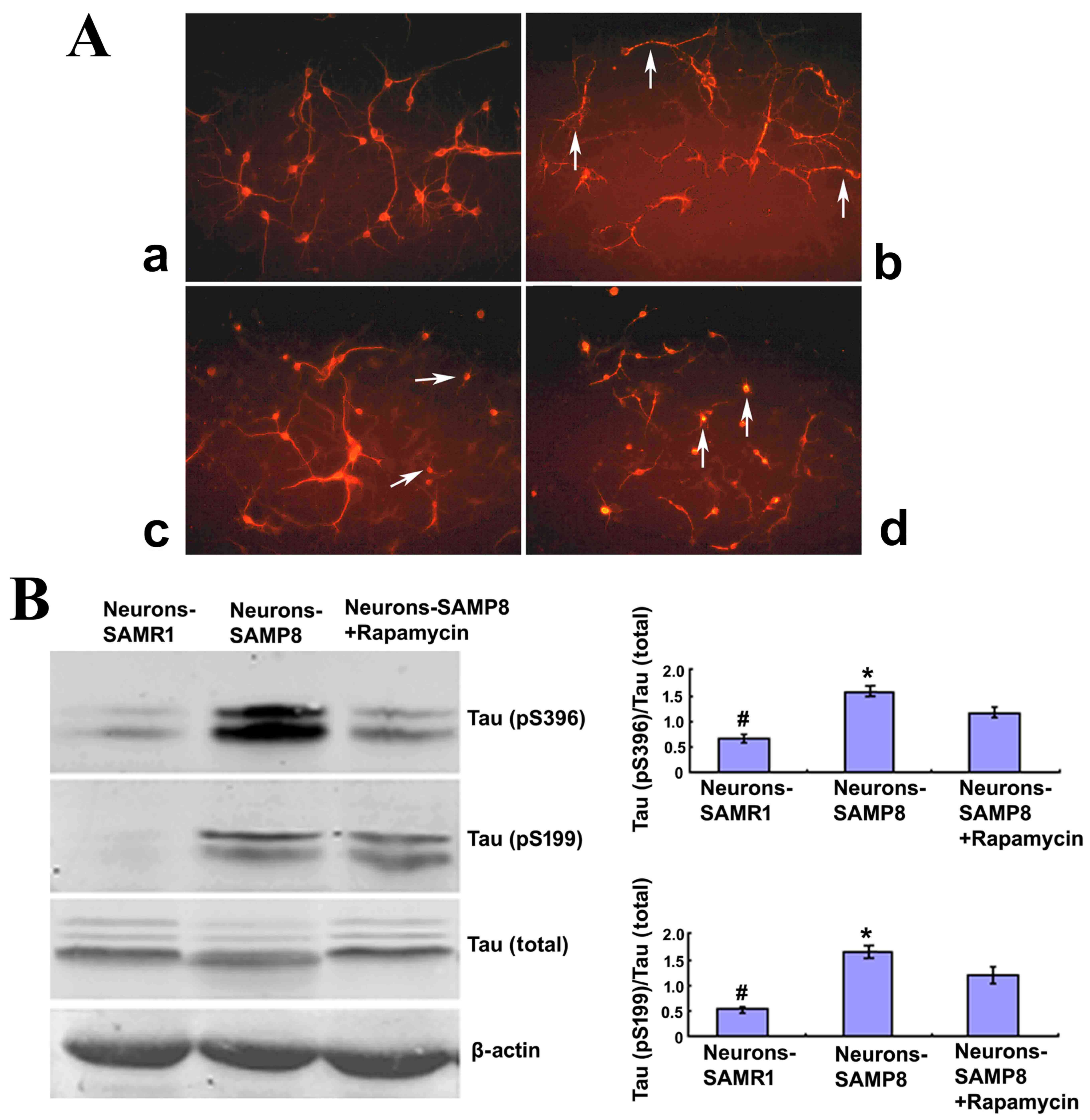 | Figure 2.Rapamycin treatment may promote cell
morphology and alleviate the Tau phosphorylation of neurons from
SAMP8. (A) Cell morphology (magnification, ×200). The neurons
derived from SAMR1 appear to be in a normal state (a,
Neurons-SAMR1). The neurons extracted from SAMP8 were in poor state
with most processes broken and exhibited ‘bead-like’ changes
(indicated by the white arrows) (b, Neurons-SAMP8). When pretreated
with 0.5 µM rapamycin, neurons from SAMP8 were partially improved
with some smooth and slender processes (indicated by the white
arrows; c, Neurons-SAMP8 + 0.5 µM rapamycin). When pretreated with
1.0 µM rapamycin, the cell state was worse than the untreated group
and most neurons lacked projections (d, Neurons-SAMP8 + 1.0 µM
rapamycin). (B) Western blot analysis was used to investigate Tau
phosphorylation. In the neurons-SAMP8 group, the protein expression
levels of Tau (pS199) and Tau (pS396) were significantly increased
when compared with the control neurons-SAMR1 group. When pretreated
with 0.5 µM rapamycin for three days, Tau (pS199) and Tau (pS396)
exhibited significantly decreased levels of protein expression when
compared with the neurons-SAMP8 group. *P<0.05 vs. neurons-SAMP8
+ Rapamycin group; #P<0.01 vs. neurons-SAMP8 group.
SAMP8, senescence accelerated mouse prone 8; SAMR1, senescence
accelerated mouse resistant 1. SAMP8 group, brain tissue of SAMP8;
SAMR1 group, brain tissue of SAMR1; neurons-SAMP8 group, neurons of
SAMP8; neurons-SAMR1 group, neurons of SAMR1. |
To further determine the effect of rapamycin
administration on cell morphology, western blot analysis was
performed to investigate the phosphorylation status of Tau, an
important microtubule-stabilizing protein in neurons (24). In the neurons-SAMP8 group, the
protein expression levels of Tau (pS199) and Tau (pS396) were
significantly increased when compared with the control
neurons-SAMR1 group (P<0.01). When pretreated with 0.5 µM
rapamycin for three days, Tau (pS199) and Tau (pS396) protein
expression levels were significantly decreased when compared with
the neurons-SAMP8 group (P<0.05; Fig.
2B). These results suggested that rapamycin treatment may
downregulate the phosphorylation of Tau protein and protect the
neurons in SAMP8 against degeneration.
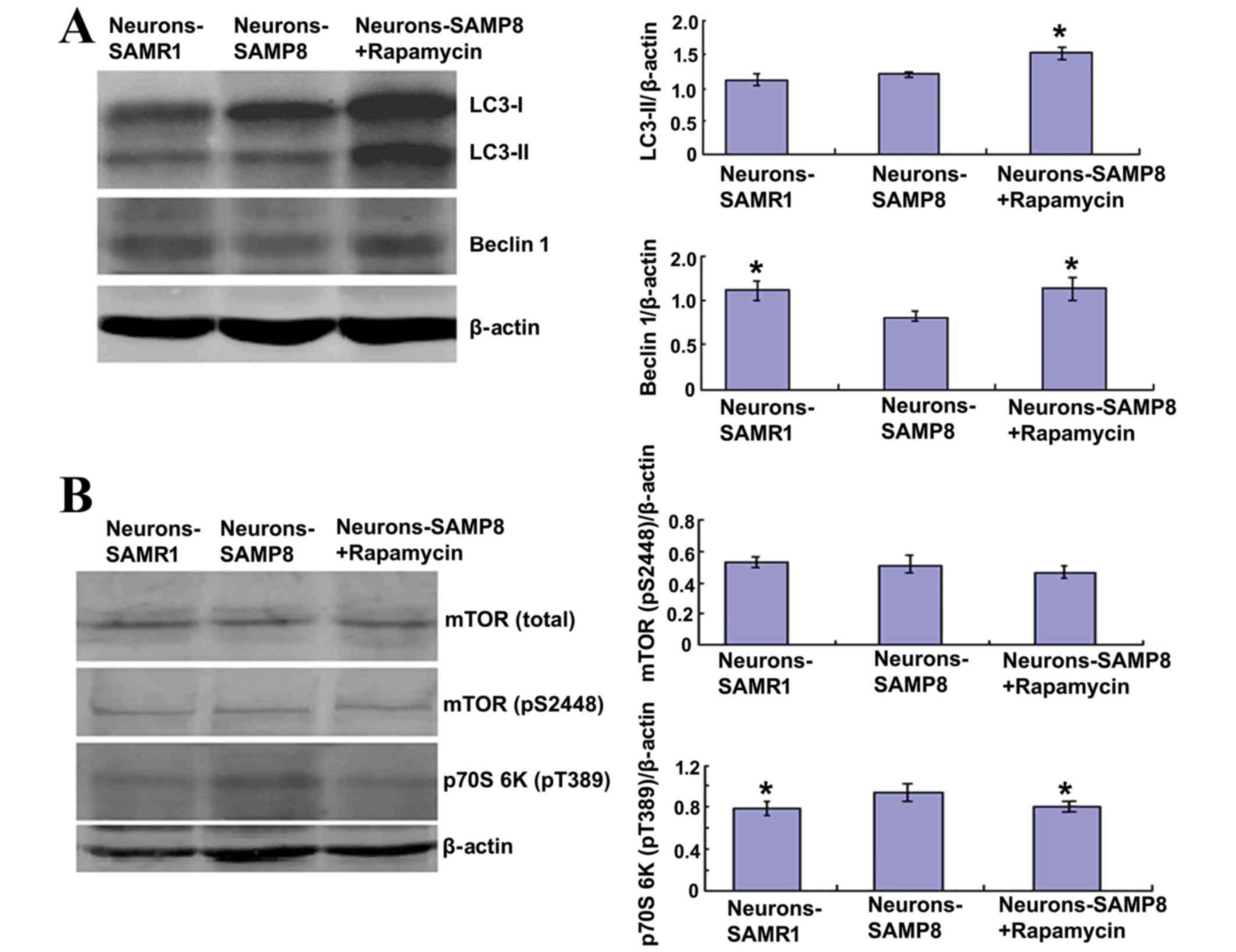
Rapamycin may inhibit mTOR signaling
and promote autophagy
Rapamycin (0.5 µM) was used to pretreat primary
neurons extracted from SAMP8. Rapamycin significantly increased the
protein expression levels of LC3-II and beclin 1 in the SAMP8
neurons (P<0.05; Fig. 3A). The
level of LC3-II is an indicator for the extent of autophagy
(25). Therefore, this result
indicated that rapamycin may stimulate autophagic activity.
Although the LC3-II protein expression levels exhibited in the
neurons-SAMR1 group was not significantly different from the
neurons in the-SAMP8 group (P>0.05), the protein expression
levels of beclin 1, a well-known key regulator of autophagy
(26), were significantly decreased
in the neurons-SAMP8 group compared with the neurons-SAMR1 group
(P<0.05; Fig. 3A). This result
indicated that the activity of autophagy was lower in the
neurons-SAMP8 at the tenth day (Fig.
3A).
The effect of 0.5 µM rapamycin on mTOR signaling was
determined using western blot analysis in primary neurons.
Rapamycin exhibited no effect on the protein expression levels of
total mTOR (P>0.05) and phospho-mTOR (P>0.05); however, the
protein expression levels of phosphorylated p70S6K at Thr389 were
significantly decreased in the rapamycin pretreated SAMP8 group
when compared with the neurons-SAMP8 group (P<0.05; Fig. 3B) which suggested that mTOR
phosphorylation is not always in accordance with its activity.
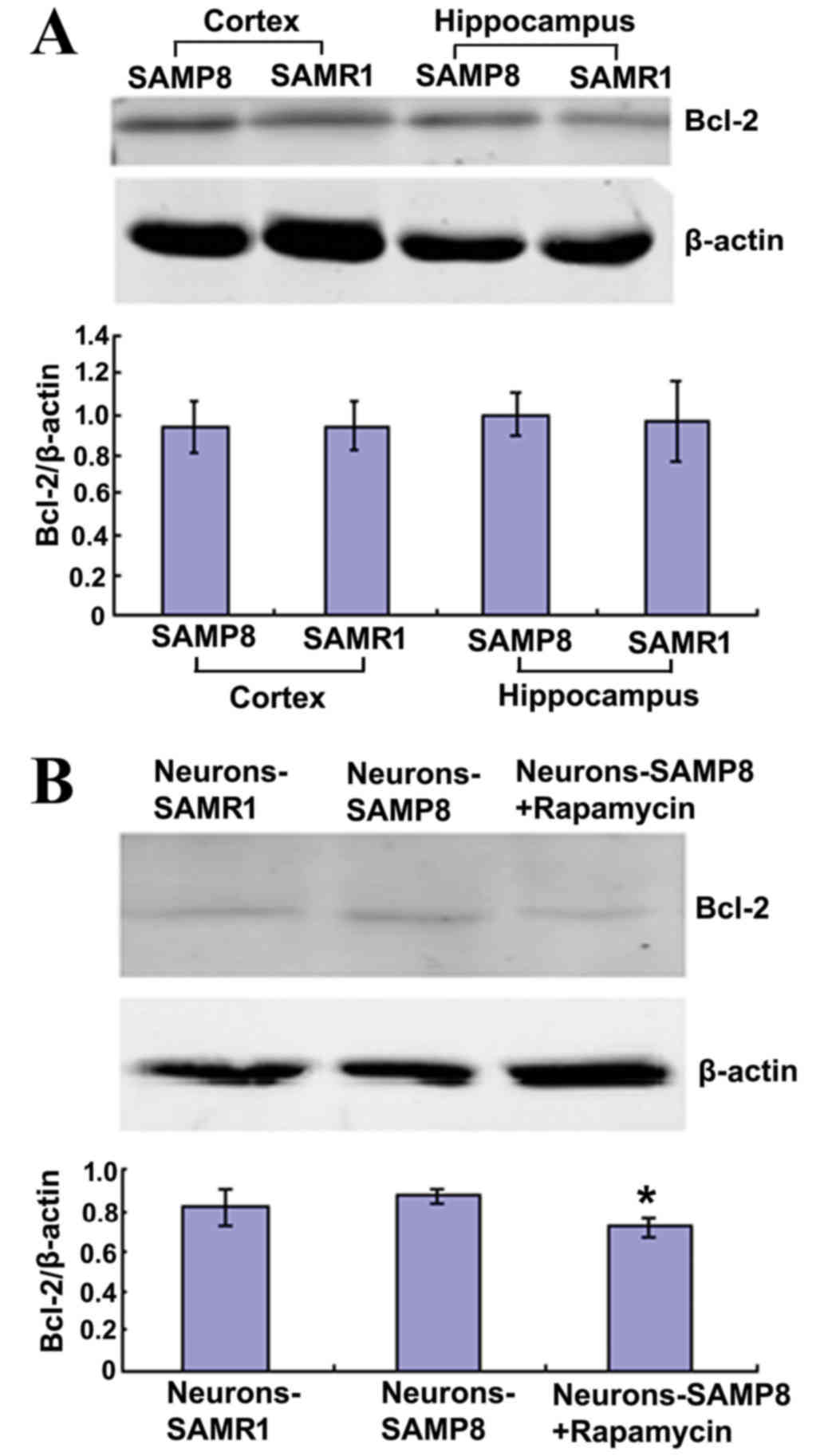
Rapamycin decreased the protein
expression level of Bcl-2 in primary neurons
Bcl-2 is an anti-apoptotic protein that is able to
inhibit macroautophagy (27). The
present findings indicated that Bcl-2 protein expression levels in
the cortex and hippocampus of 12-month-old SAMP8 mice exhibited no
difference when compared with matched SAMR1 mice and the primary
neurons from the two strains (P>0.05; Fig. 4A). However, in the neurons-SAMP8 +
rapamycin group, Bcl-2 protein expression levels were significantly
decreased when compared with the neurons-SAMP8 group (P<0.05;
Fig. 4B).
Discussion
The major findings of the present study indicated
that the levels of mTOR (pSer2448) and phosphorylated p70S6K at
Thr389 were significantly increased in SAMP8 hippocampal neurons
when compared with the control SAMR1 group. Furthermore, mTOR
activity signaling was upregulated in the neurons of SAMP8. The
increase of mTOR signaling in the hippocampus and primary neurons
of aged SAMP8 mice may contribute to neurodegenerative disorders in
SAMP8.
Rapamycin is a specific inhibitor of mTOR, which has
been used in vitro and in vivo to block mTOR function
(28). The present results
demonstrated that rapamycin administration significantly increased
the protein expression levels of phosphorylated p70S6K in the SAMP8
group when compared with the SAMR1 group. When treated with 0.5 µM
rapamycin for three days, the appearance of neurons from the
neurons-SAMP8 group were improved and Tau (pS199) and Tau (pS396)
protein expression levels were decreased when compared with the
untreated neurons-SAMP8 group. Western blot analysis of brain
lysates from rapamycin-treated and the control SAMR1 groups showed
a significant decrease in the phosphorylation of p70S6K,
demonstrating the effective inhibition of rapamycin on the mTOR
pathway. Previous data from studies conducted on Alzheimer's
disease brains have further strengthened the link of mTOR and Tau
and demonstrated that mTOR signaling is selectively increased in
neurons predicted to develop neurofibrillary tangles and such an
increase correlated with Tau phosphorylation (21,29,30). The
present results suggest that rapamycin may provide neuron
protection through the inhibition of mTOR and alleviation of Tau
phosphorylation.
The autophagy process recycles unnecessary or
damaged material, therefore, not only providing nutrients to
maintain vital cellular functions in times of starvation but also
eliminating potentially harmful cellular material (31). Autophagy is constitutively active in
healthy neurons (32,33). A previous study using neuron-specific
autophagy protein-deficient mice (atg5−/− or
atg7−/−) exhibited abnormal protein aggregates,
neurodegeneration and subsequent motor dysfunction, which suggested
that autophagy is essential for neuronal homeostasis and quality
control (34). Furthermore, a
previous study demonstrated a correlation with the cognitive
decline in the hippocampal neurons of 12-month-old SAMP8 mice with
an increase in ubiquitinated proteins (35). mTOR is able to control protein
turnover though inhibiting autophagy (36); therefore, the basal and optimal
levels of autophagy are essential for neurons to efficiently remove
damaged organelles and misfolded proteins.
Bcl-2 is an anti-apoptotic protein that is able to
inhibit autophagy (37). In the
present study, when rapamycin was administered, the Bcl-2 protein
expression levels declined significantly in the neurons-SAMP8
group, indicating that Bcl-2 may be regulated by rapamycin through
mTOR signaling. However, no difference was indicated in the Bcl-2
protein expression levels in the cortex and hippocampus of
12-month-old SAMP8 mice when compared with the matched SAMR1 mice
and the primary neurons from the two strains, indicating that Bcl-2
may not participate in the aging process of SAMP8. Results from
previous studies have demonstrated that beclin 1 is an important
component of the phosphoinositide 3-kinase complex that regulates
autophagosome maturation, endocytic trafficking and the
downregulation of beclin 1 decreases the activity of autophagy
(37,38). Further studies have revealed that
starvation induces Jun N-terminal kinase 1 activity, which may
phosphorylate Bcl-2, thereby disrupting the association between
beclin 1 and Bcl-2 to induce autophagy (39). In an alternative study (40), LC3-II and beclin 1 expression levels
were increased in brain areas of seven-month-old SAMP8; At 12
months, LC3-II expression remained increased whereas beclin 1
expression was diminished, suggesting that autophagic activity may
increase reactively at the beginning of Alzheimer's disease and
decline with aging. The pathological changes of 12-month-old SAMP8
are similar to late-onset Alzheimer's disease from the perspective
of autophagy (41).
In conclusion, the present study demonstrated that
mTOR signaling plays a key role in the neurodegenerative process,
and rapamycin administration could protect neurons and alleviate
Tau phosphorylation of SAMP8 mice. In addition, rapamycin might
have a potential role in reducing cognitive decline. However, the
current study of rapamycin was limited to in vitro results.
Thus, further in vivo studies in animal models are
required.
Acknowledgements
The present study was supported by the Hebei
Department of Health (Grant No ZL20140075; Hebei, China). The
authors acknowledge the expert technical assistance of Dr Lingling
Jiang, and the donation of SAMP8 and SAMR1 mice from Professor
David Yew.
References
|
1
|
Keith CT and Schreiber SL: PIK-related
kinases: DNA repair, recombination, and cell cycle checkpoints.
Science. 270:50–51. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takei N and Nawa H: mTOR signaling and its
roles in normal and abnormal brain development. Front Mol Neurosci.
7:282014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sarkar S: Regulation of autophagy by
mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction
in neurodegenerative diseases and therapeutic application of
autophagy enhancers. Biochem Soc Trans. 41:1103–1130. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mariño G, Madeo F and Kroemer G: Autophagy
for tissue homeostasis and neuroprotection. Curr Opin Cell Biol.
23:198–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rubinsztein DC: The roles of intracellular
protein-degradation pathways in neurodegeneration. Nature.
443:780–786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan T, Kondo S, Le W and Jankovic J: The
role of autophagy-lysosome pathway in neurodegeneration associated
with Parkinson's disease. Brain. 131:1969–1978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rivero-Ríos P, Madero-Pérez J, Fernández B
and Hilfiker S: Targeting the Autophagy/Lysosomal Degradation
Pathway in Parkinson's Disease. Curr Neuropharmacol. 14:238–249.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wong M: Mammalian target of rapamycin
(mTOR) pathways in neurological diseases. Biomed J. 36:40–50. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ganley IG, Lam du H, Wang J, Ding X, Chen
S and Jiang X: ULK1.ATG13.FIP200 complex mediates mTOR signaling
and is essential for autophagy. J Biol Chem. 284:12297–12305. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen J, Zheng XF, Brown EJ and Schreiber
SL: Identification of an 11-kDa FKBP12-rapamycin-binding domain
within the 289-kDa FKBP12-rapamycin-associated protein and
characterization of a critical serine residue. Proc Natl Acad Sci
USA. 92:pp. 4947–4951. 1995; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi J, Chen J, Schreiber SL and Clardy J:
Structure of the FKBP12-rapamycin complex interacting with the
binding domain of human FRAP. Science. 273:239–242. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
King MA, Hands S, Hafiz F, Mizushima N,
Tolkovsky AM and Wyttenbach A: Rapamycin inhibits polyglutamine
aggregation independently of autophagy by reducing protein
synthesis. Mol Pharmacol. 73:1052–1063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tóth ML, Sigmond T, Borsos E, Barna J,
Erdélyi P, Takács-Vellai K, Orosz L, Kovács AL, Csikós G, Sass M
and Vellai T: Longevity pathways converge on autophagy genes to
regulate life span in Caenorhabditis elegans. Autophagy. 4:330–338.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harrison DE, Strong R, Sharp ZD, Nelson
JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter
CS, et al: Rapamycin fed late in life extends lifespan in
genetically heterogeneous mice. Nature. 460:392–395.
2009.PubMed/NCBI
|
|
20
|
Miller RA, Harrison DE, Astle CM, Baur JA,
Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF,
et al: Rapamycin, but not resveratrol or simvastatin, extends life
span of genetically heterogeneous mice. J Gerontol A Biol Sci Med
Sci. 66:191–201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pei JJ, Björkdahl C, Zhang H, Zhou X and
Winblad B: p70 S6 kinase and tau in Alzheimer's disease. J
Alzheimers Dis. 14:385–392. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Das F, Ghosh-Choudhury N, Mahimainathan L,
Venkatesan B, Feliers D, Riley DJ, Kasinath BS and Choudhury GG:
Raptor-rictor axis in TGFbeta-induced protein synthesis. Cell
Signal. 20:409–423. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meske V, Albert F and Ohm TG: Coupling of
mammalian target of rapamycin with phosphoinositide 3-kinase
signaling pathway regulates protein phosphatase 2A- and glycogen
synthase kinase-3-dependent phosphorylation of Tau. J Biol Chem.
283:100–109. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barth S, Glick D and Macleod KF:
Autophagy: Assays and artifacts. J Pathol. 221:117–124. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang W, Li Q, Song C and Lao L: Knockdown
of autophagy-related protein 6, Beclin-1, decreases cell growth,
invasion, and metastasis and has a positive effect on
chemotherapy-induced cytotoxicity in osteosarcoma cells. Tumour
Biol. 36:2531–2539. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dancey JE: Inhibitors of the mammalian
target of rapamycin. Expert Opin Investig Drugs. 14:313–328. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
An WL, Cowburn RF, Li L, Braak H,
Alafuzoff I, Iqbal K, Iqbal IG, Winblad B and Pei JJ: Up-regulation
of phosphorylated/activated p70 S6 kinase and its relationship to
neurofibrillary pathology in Alzheimer's disease. Am J Pathol.
163:591–607. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pei JJ and Hugon J: mTOR-dependent
signalling in Alzheimer's disease. J Cell Mol Med. 12:2525–2532.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee JA: Neuronal autophagy: A housekeeper
or a fighter in neuronal cell survival? Exp Neurobiol. 21:1–8.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xilouri M and Stefanis L: Autophagy in the
central nervous system: Implications for neurodegenerative
disorders. CNS Neurol Disord Drug Targets. 9:701–719. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mizushima N and Levine B: Autophagy in
mammalian development and differentiation. Nat Cell Biol.
12:823–830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma Q, Qiang J, Gu P, Wang Y, Geng Y and
Wang M: Age-related autophagy alterations in the brain of
senescence accelerated mouse prone 8 (SAMP8) mice. Exp Gerontol.
46:533–541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Díaz-Troya S, Pérez-Pérez ME, Florencio FJ
and Crespo JL: The role of TOR in autophagy regulation from yeast
to plants and mammals. Autophagy. 4:851–865. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klionsky DJ, Codogno P, Cuervo AM, Deretic
V, Elazar Z, Fueyo-Margareto J, Gewirtz DA, Kroemer G, Levine B,
Mizushima N, et al: A comprehensive glossary of autophagy-related
molecules and processes. Autophagy. 6:438–448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Diwan A, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Menardo J, Tang Y, Ladrech S, Lenoir M,
Casas F, Michel C, Bourien J, Ruel J, Rebillard G, Maurice T, et
al: Oxidative stress, inflammation, and autophagic stress as the
key mechanisms of premature age-related hearing loss in SAMP8 mouse
Cochlea. Antioxid Redox Signal. 16:263–274. 2012. View Article : Google Scholar : PubMed/NCBI
|