Introduction
Atherosclerosis (AS), one of the most common
cardiovascular diseases, is a major cause of morbidity and
mortality worldwide (1).
Hyperlipidemia, monocyte recruitment, differentiation into
macrophages, foam cell formation and induced inflammation are the
key cellular events of AS (2).
Chronic inflammation has a key role in the occurrence and
development of AS (3), and
macrophages with the ability to stimulate the vascular inflammatory
reaction are the major effector cells throughout the pathological
process of AS (4). Therefore, genes
and cytokines involved in immune system control are essential in
regulating AS. Recent studies have indicated the potential roles of
microRNAs (miRNAs) in the regulation of AS-associated processes
(5,6).
miRNAs are small (~22 nucleotides in length),
endogenous, non-coding, single-stranded RNAs, which have important
roles in gene expression regulation by binding to the
3′-untranslated region (UTR) of target mRNAs (7,8). A
growing body of evidence suggested that miRNAs have different roles
in the development of AS. Li et al (9) reported that increased miR-155 relieves
chronic inflammation in AS. Xu et al (10) suggested that miR-135b-5p and
miR-499a-3p promoted cell proliferation and migration in AS. Zhang
et al (11) demonstrated that
miR-26a prevented endothelial cell apoptosis under AS conditions.
Ouimet et al (12) found that
miR-33 antagonism exerted atheroprotective roles via regulating
macrophage-associated inflammation.
A previous study demonstrated upregulation of
miR-146b-5p in oxidized low-density lipoprotein (oxLDL)-stimulated
monocytes (13); however, to the
best of our knowledge, no further study on the role of miR-146b-5p
in AS has been performed. Thus, the present study investigated the
potential role of miR-146b-5p in AS and explored the underlying
molecular mechanisms.
Materials and methods
Specimens
A total of 10 pairs of atherosclerotic lesion
tissues and normal veins were identified and collected during
biopsies from 10 patients (gender ratio: 1:1; age, ranging from 42
to 59 years old) with AS who were diagnosed by clinical symptoms
and angiography at the Affiliated Hospital of Qingdao University
(Qingdao, China) between August 2015 and August 2016. The exclusion
criteria of the patients were as previously described (9). Informed consent was obtained from each
patient. The present study was approved by the Affiliated Hospital
of Qingdao University (Qingdao, China).
Cell culture and foam cell model
construction
The THP-1 human monocytic and the HEK293T human
embryonic kidney cell line were obtained from the American Type
Culture Collection (Manassas, VA, USA). THP-1 cells were grown in
RPMI-1640 medium, supplemented with 10% fetal bovine serum (both
from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA USA), and
1% streptomycin and penicillin mixed solution (Thermo Fisher
Scientific, Inc.). HEK-293T cells were cultured in Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.).
All cells were incubated in a humidified atmosphere with 5%
CO2 at 37°C. A foam cell model was established as
previously described (14–16). In brief, THP-1 cells were first
seeded in culture plates at 1×106 cells/ml with 100 nM
phorbol 12-myristate 13-acetate (PMA; Sangon Biotech Co., Ltd.,
Shanghai, China) for 12 h to differentiate them into macrophages.
Subsequently, the cells were stimulated with different
concentrations of oxLDL (10, 50 or 100 µg/ml; Sangon Biotech Co.,
Ltd.) for specific durations (0, 6 or 12 h) in order to induce foam
cell formation; control cells were treated with PBS.
Oil Red O staining
Macrophages derived from THP-1 cells were
transfected with a miR-146b-5p inhibitor or its negative control
(GenScript Biotech Corporation; Piscataway, NJ, USA) using
Lipofectamine 2000 transfection reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Following transfection with miR-146b-5p
inhibitor or control, cells were treated with oxidized low-density
lipoprotein (50 µg/ml) for 24 h. The cells were then washed with
PBS, fixed with 4% paraformaldehyde, stained with Oil Red O (Sangon
Biotech Co., Ltd.) at room temperature for 20 min, and then
de-stained with 60% isopropanol for 1 min. The foam cells were then
imaged by using a microscope (Olympus Corporation,, Tokyo, Japan)
at a magnification of ×40.
RNA isolation and
reverse-transcription quantitative polymerase chain reaction
(RT-qPCR)
miR-146b-5p was extracted from atherosclerotic
lesion tissues and normal veins by using the mirVana PARIS kit (cat
no. AM1556; Ambion; Thermo Fisher Scientific, Inc.) in line with
the manufacturer's instructions. The TaqMan MicroRNA Reverse
Transcription kit (cat no. 4366596; Applied Biosystems; Thermo
Fisher Scientific, Inc.) was used for RT of miRNA, and the
complementary (c)DNA was amplified by PCR using TaqMan Fast
Advanced Master Mix (cat no. 4444556; Applied Biosystems; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The amplification conditions were as follows: 37
cycles of denaturation at 95°C for 10 sec, followed by annealing
and extension at 58°C for 60 sec. U6 was used as an endogenous
control of miRNA expression. The primer sequences were as follows:
miR-146b-5p forward, 5′-TGACCCATCCTGGGCCTCAA-3′ and reverse,
5′-CCAGTGGGCAAGATGTGGGCC-3′; and U6 forward,
5′GCTTCGGCAGCACATATACTAAAAT3′ and reverse,
5′CGCTTCACGAATTTGCGTGTCAT3′.
For tumor necrosis factor (TNF) receptor-associated
factor 6 (TRAF6) mRNA expression analysis, total RNA from
THP-1-derived macrophages was isolated by using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was obtained by
using a RT-PCR detection kit (cat no. 18091050; Invitrogen; Thermo
Fisher Scientific, Inc.) following the manufacturer's instructions.
Real-time PCR was performed to amplify the synthesized cDNA by
using the Fast SYBR Green Master Mix (cat no. 4385610; Applied
Biosystems; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Amplification conditions were 95°C for
10 min, followed by 38 cycles at 95°C for 15 sec, and 72°C for 30
sec. GAPDH acted as an internal control. The 2−ΔΔCq
method was used to calculate the relative gene expression (17). The primer sequences were as follows:
TRAF6 forward, 5′-GAGTTTGACCCACCTCTGGA-3′ and reverse,
5′-TTTCATTGTCAACTGGGCACT-3′; GAPDH forward,
5′-CTTTGGTATCGTGGAAGGACTC-3′ and reverse,
5′-GTAGAGGCAGGGATGATGTTCT-3′.
Western blot analysis
Cells were dissolved by using
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology, Haimen, China) and a bicinchoninic acid protein
assay kit (cat no. 23225; Thermo Fisher Scientific, Inc.) was used
for determining the protein concentration. Protein samples (25 µg
per lane) were separated by 10% SDS-PAGE, transferred to a
polyvinylidene difluoride membrane and blocked with 5% skimmed milk
powder. The membrane was incubated with a primary antibody against
GAPDH (cat no. 5174), nuclear factor (NF)-κB (p65; cat no. 8214) or
TRAF6 (cat no. 8028) (dilution for all, 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA) overnight at 4°C, and then
incubated with anti-rabbit IgG horseradish peroxidase-conjugated
secondary antibody (cat no. 7074; 1:5,000 dilution; Cell Signaling
Technology, Inc.) at room temperature for 2 h. The protein levels
were detected by enhanced chemiluminescence using the
Chemiluminescent ECL reagent (EMD Millipore, Billerica, MA,
USA).
ELISA
Pro-inflammatory cytokines [interleukin (IL)-6, cat
no. E-EL-H0102c; Elabscience Biotechnology, Inc., Wuhan, China),
IL-2 (cat no. 8629), TNF-α (cat no. 8668) (both from Cell Signaling
Technology, Inc.) and transforming growth factor (TGF)-β (cat no.
BMS249-4FIVE; Invitrogen; Thermo Fisher Scientific, Inc.) secreted
from THP-1-derived macrophages transfected with miR-146b-5p
inhibitor or control, followed by treatment with oxidized
low-density lipoprotein (50 µg/ml) for 24 h, were detected by using
respective ELISA kits according to the manufacturer's
instructions.
Luciferase reporter assay
To predict the potential targets of miR-146b-5p,
TargetScan (http://www.targetscan.org/vert_71/) was performed in
the present study. To confirm our prediction, miR-146b-5p
recognition sequences from the wild-type (WT) and mutant (MUT)
3′-UTR of TRAF6 (forward, 5′-GCGATCGCTATATGTAATATATTAAAAGTGAAA-3′
and reverse, 5′-GGAGCTCAAATAATTAAGGTTATATTTAGG-3′) were amplified
and then cloned into the psiCHECK-2 reporter vector (Promega
Corporation, Madison, WI, USA). The resulting
miR-146b-5p-TRAF6-3′UTR-WT or miR-146b-5p-TRAF6-3′UTR-MUT vectors
were respectively co-transfected with miR-146b-5p or control miR
into HEK293T cells using Lipofectamine 2000 transfection reagent in
line with the manufacturer's protocol. At 48 h after transfection,
the Dual-Luciferase Reporter Assay system (Promega Corporation) was
used for luciferase activity detection. The luciferase activity was
then normalized, expressed and Renilla luciferase activity was used
as the internal control.
Statistical analysis
Data are presented as the mean ± standard deviation.
Student's t-test was performed for comparison between two groups.
SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA) was used for
statistical analysis. All tests were independently performed at
least three times. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-146b-5p is overexpressed in the
atherosclerotic lesions of patients with AS and is induced by oxLDL
in human macrophages
The relative expression levels of miR146b-5p in
atherosclerotic lesions and normal veins from the same AS patients
were detected by RT-qPCR, and the results demonstrated that the
miR146b-5p expression levels were significantly increased in the
atherosclerotic lesions compared with those in the normal veins. To
determine whether overexpression of miR-146b-5p is induced by
oxLDL, THP-1 cells were first treated with 100 nM PMA to stimulate
their differentiation into macrophages and then stimulated with
various concentrations of oxLDL (0, 10, 50 and 100 µg/ml) for 24 h
or with 50 µg/ml oxLDL for specific durations (0, 6 or 12 h) in
order to induce foam cell formation (18). The findings demonstrated that the
expression of miR-146b-5p was significantly increased by oxLDL
stimulation, and the effect was dose- and time-dependent (Fig. 1).
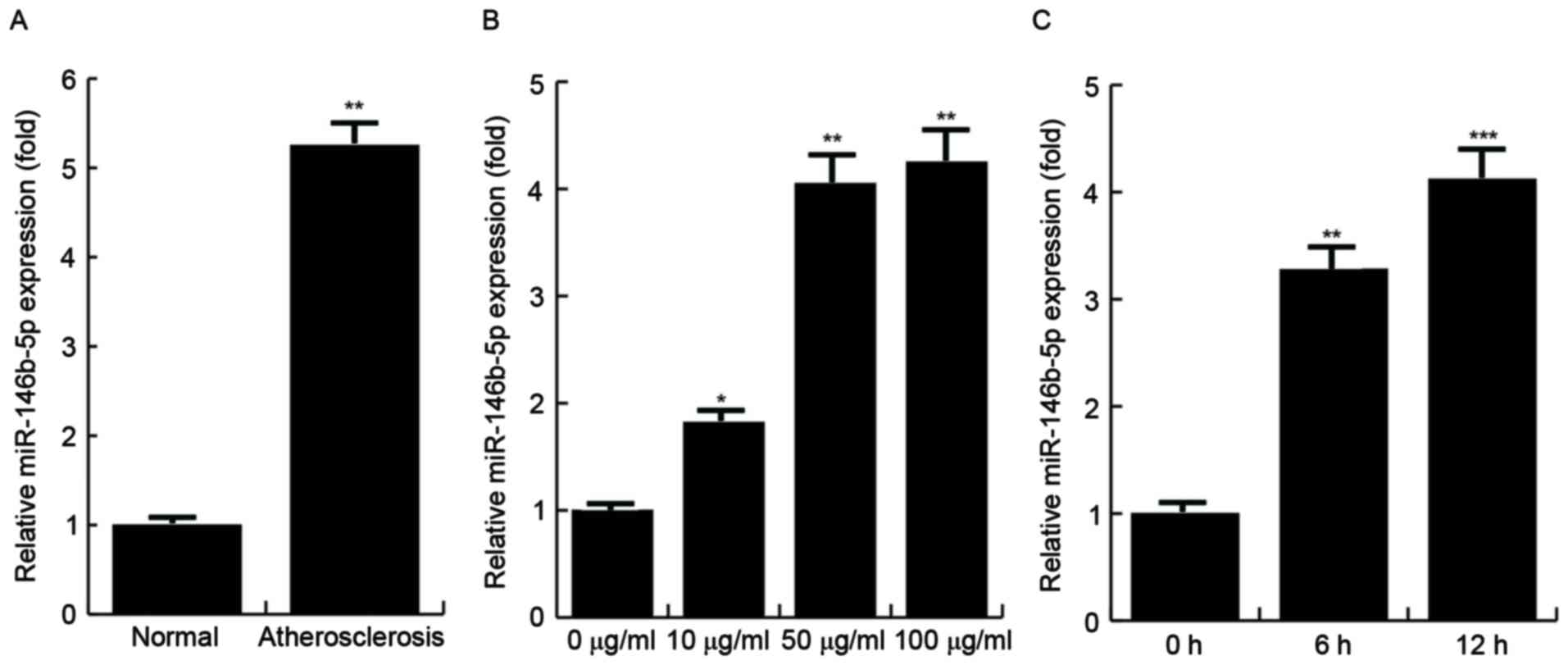 | Figure 1.Expression of miR-146b-5p is increased
in human macrophages in clinical specimens and is induced by oxLDL.
(A) Levels of miR-146b-5p in atherosclerotic lesions (n=10) and
normal veins (n=10) from the same patients with AS. (B) Analysis of
miR-146b-5p expression in THP-1 cells, which were first stimulated
with PMA (100 nM) to induce them to differentiate into macrophages
and then treated with oxLDL at the indicated doses (0, 10, 50 or
100 µg/ml). (C) miR-146b-5p expression in THP-1 cells, which were
first stimulated with PMA (100 nM) to induce them to differentiate
into macrophages and then treated with oxLDL (50 µg/ml) for the
indicated times (0, 6 or 12 h). miR-146b-5p was determined by
reverse-transcription quantitative polymerase chain reaction
analysis. *P<0.05, **P<0.01, ***P<0.001 vs. control. miR,
microRNA; oxLDL, oxidized low-density lipoprotein; PMA, phorbol
12-myristate 13-acetate. |
TRAF6 is a functional target of
miR-146b-5p
To explore the mechanisms by which miR-146b-5p
affects macrophage-derived foam cell formation and the inflammatory
response, TargetScan and miRanda database searches were performed
to predict the direct mRNA targets of miR-146b-5p. To confirm the
prediction of TRAF6 mRNA being a target, a luciferase reporter
assay was performed. The results revealed that the luciferase
activity was significantly decreased in the HEK293 cells
co-transfected with miR-146b-5p and miR-146b-5p-TRAF6-WT, while
co-transfection of miR-146b-5p with miR-146b-5p-TRAF6-MUT did not
(Fig. 2). This result indicated that
TRAF6 is a direct target of miR-146b-5p.
miR-146b-5p inhibition during foam
cell formation increases TRAF6
To investigate the role of miR-146b-5p in the
pathological process of AS, miR-146b-5p was inhibited using
miR-146b-5p inhibitor. THP-1 cells, which had been treated with 100
nM PMA for 12 h to induce their differentiation into macrophages,
were transfected with miR-146b-5p inhibitor or its negative control
for 24 h, and then treated with 50 µg/ml oxLDL for 24 hto induce
foam cell formation. Efficient inhibition of miR-146b-5p was
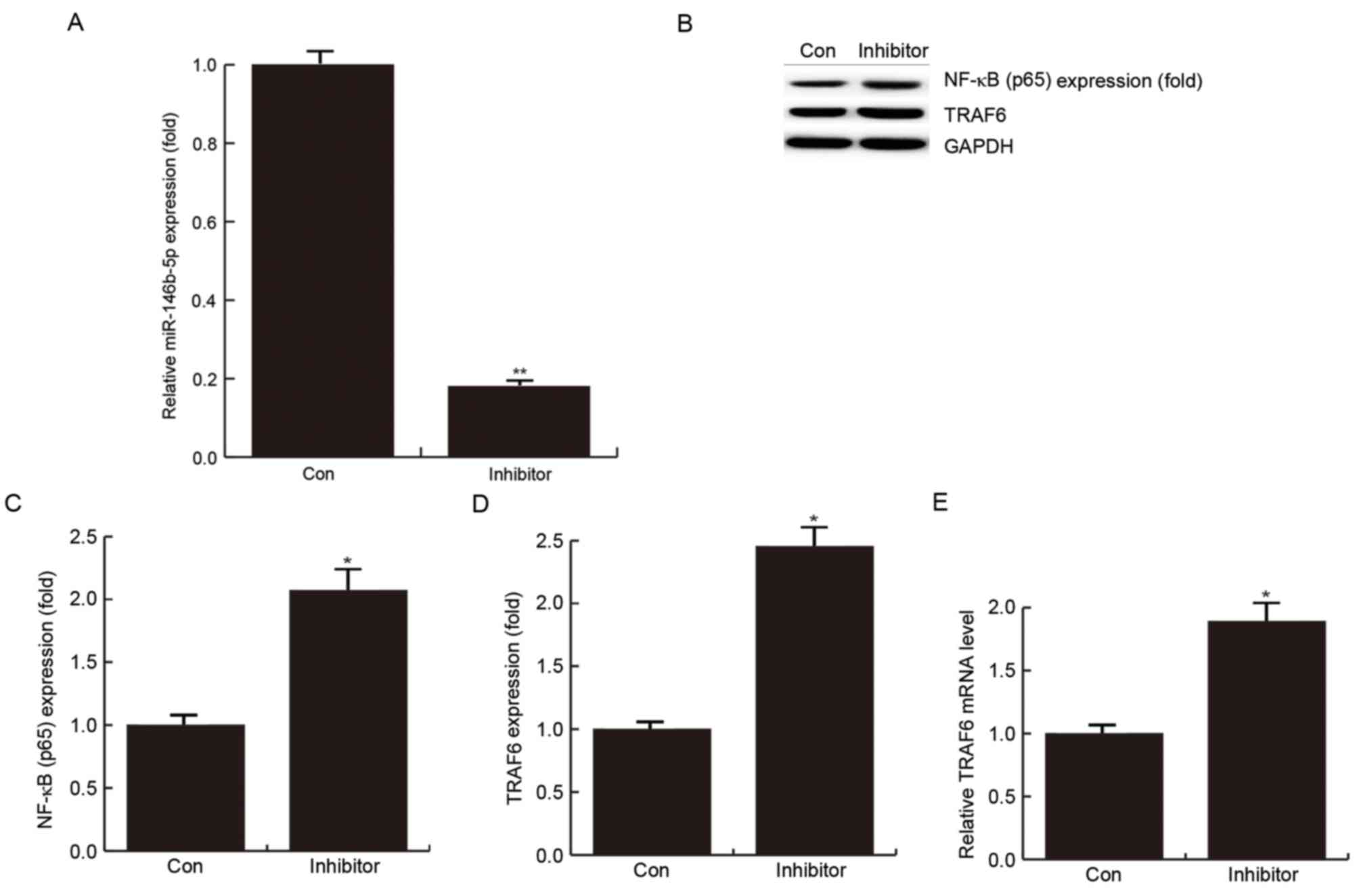
confirmed by RT-qPCR (Fig. 3A).
In addition, western blot analysis demonstrated that
after transfection with miR-146b-5p inhibitor for 24 h, the protein
levels of NF-κB (p65) (P<0.05; Fig.
3B and C) and TRAF6 were significantly enhanced (P<0.05;
Fig. 3B and D). The results also
indicated that miR-146b-5p inhibition may promote the inflammatory
response, possibly by promoting the expression of NF-κB (p65) in
foam cell formation. At the mRNA level, TRAF6 was significantly
enhanced in the group transfected with miR-146b-5p inhibitor
(Fig. 3E). This further supported
the result that TRAF6 is a direct target of miR-146b-5p.
miR-146b-5p inhibition promotes the
inflammatory response during foam cell formation
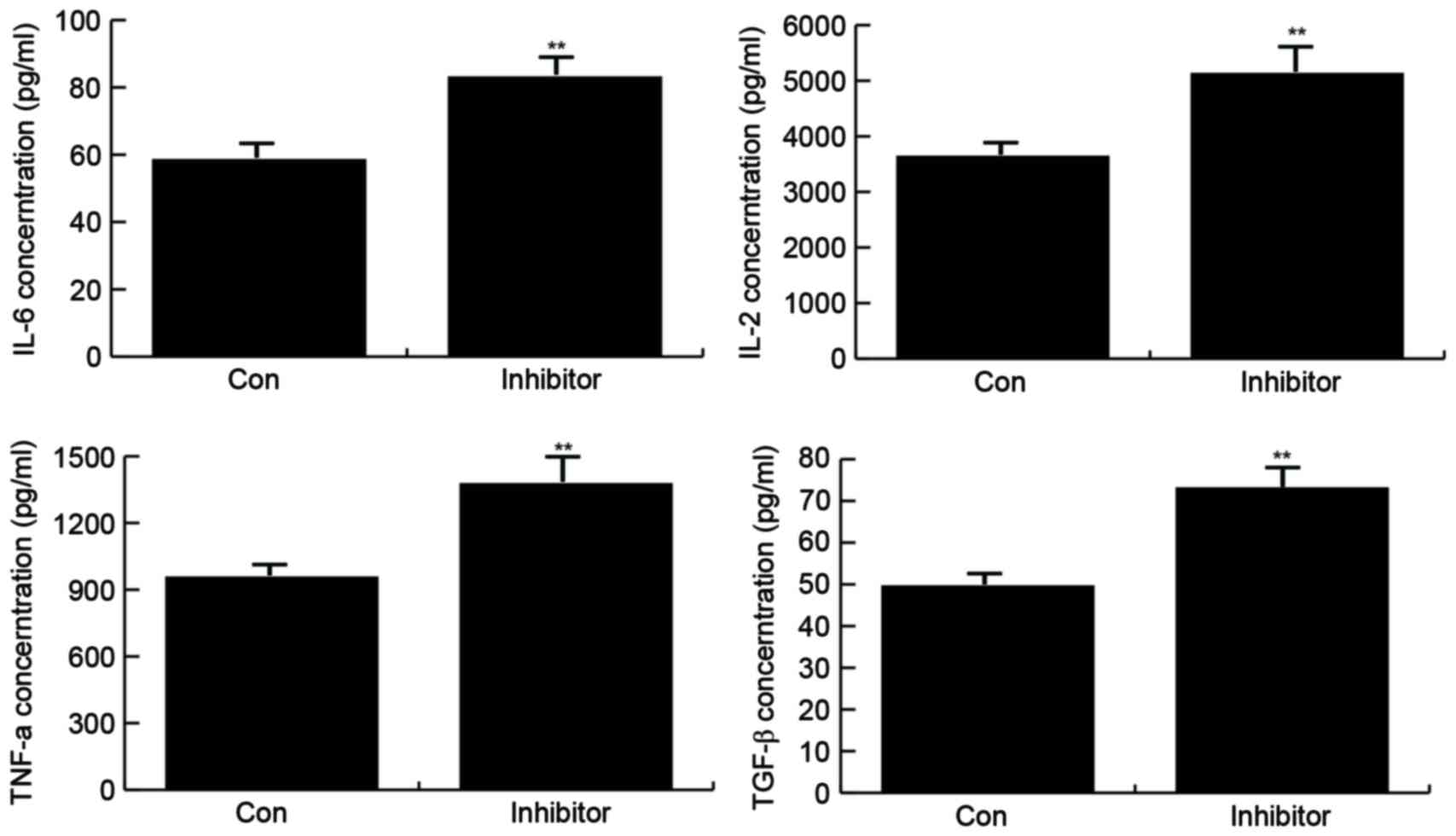
The effect of miR-146b-5p inhibition on inflammatory
factor secretion in macrophages treated with oxidized low-density
lipoprotein (50 µg/ml) for 24 h was examined by ELISA. The results
demonstrated that in the group transfected with miR-146b-5p
inhibitor, the secretion of IL-6, IL-2, TNF-α and TGF-β was
significantly increased (Fig.
4).
miR-146b-5p inhibition enhances lipid
uptake during foam cell formation
The effect of miR-146b-5p inhibition on foam cell
formation was analyzed by Oil Red O staining. The results indicated
that in the group transfected with miR-146b-5p, lipid uptake was
notably enhanced (Fig. 5).
Discussion
Increasing evidence suggested that chronic
inflammation contributes to the formation of atherosclerotic
lesions. Foam cell formation may be induced by exposure of
macrophages to oxLDL (19).
Exploration of the potential molecular mechanisms of inflammatory
processes and foam cell formation will provide novel strategies for
the treatment of AS.
Various studies have demonstrated that miRNAs have
an important role in the pathogenesis of AS. Zernecke et al
(20) demonstrated that miR-126
prevents atherosclerotic lesion formation through regulating
angiogenesis and vascular inflammation, and an anti-AS function of
miR-126-5p has been reported recently (21). Consistent with these results, Tabet
et al (22) suggested that
the anti-inflammatory function of high-density lipoprotein is
conferred through miR-223. By contrast, Zhang et al
(23) reported that miR-150 promotes
AS via enhancing endothelial cell migration. The present study
investigated the role of miR-146b-5p in AS-associated processes to
identify novel therapeutic strategies for the treatment of AS and
other vascular diseases.
First, the expression levels of miR-146b-5p were
determined in atherosclerotic lesions of patients with AS, and the
results indicated that miR-146b-5p was highly expressed in the
atherosclerotic lesions compared with that in normal veins from the
same patient. The present study also found that miR-146b-5p may be
induced by oxLDL stimulation in a time- and dose-dependent manner,
which was consistent with the results of a previous study (13).
Next, the present study confirmed TRAF6 as a direct
target of miR-146b-5p. A statistically significant inverse
association was identified between miR-146b-5p and TRAF6 expression
in oxLDL-stimulated macrophages, suggesting a significant
biological function of the TRAF6-miR-146b-5p complex in AS. TRAF6
functions as a signal transducer in the NF-κB pathway, and the
activation of NF-κB was reported to be elevated in patients with
acute coronary syndrome and in oxLDL-induced mast cells (24,25).
Various molecules involved in the immune response and early
inflammation are modulated by the NF-κB pathway. Thus, it was
hypothesized that TRAF6-miR-146b-5p is involved in the regulation
of AS-associated inflammation. The results of the present study
suggested that inhibition of miR146b-5p increases TRAF6 and NF-κB
(p65) expression, as well as the secretion of pro-inflammatory
cytokines (IL-6, IL-2, TNF-α and TGF-β) in oxLDL-simulated
macrophages. These results confirmed the prediction that
miR-146b-5p acts as a promoter of inflammation in oxLDL-stimulated
macrophages, partly via targeting TRAF6. Furthermore, the present
study investigated the effect of miR-146b-5p on foam cell formation
via Oil Red O staining. The results indicated that miR-146b-5p
inhibition significantly enhanced the lipid uptake by
oxLDL-stimulated macrophages.
In conclusion, the present study found that
miR146b-5p is overexpressed in the atherosclerotic lesions of
patients with AS, and it may be induced by oxLDL in human
macrophages. Blockade of miR146b-5p promoted inflammation and foam
cell formation by increasing TRAF6-mediated activation of NF-κB
expression, indicating the anti-AS function of miR146b-5p.
References
|
1
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Imanishi T and Akasaka T: Novel strategies
to target inflammatory processes in atherosclerosis. Curr Pharm
Des. 19:1616–1625. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moore KJ and Tabas I: Macrophages in the
pathogenesis of atherosclerosis. Cell. 145:341–355. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang C: MicroRNAs in vascular biology and
vascular disease. J Cardiovasc Transl Res. 3:235–240. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang C: MicroRNAs: Role in cardiovascular
biology and disease. Clin Sci (Lond). 114:699–706. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ambros V: microRNAs: Tiny regulators with
great potential. Cell. 107:823–826. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eulalio A, Huntzinger E and Izaurralde E:
Getting to the root of miRNA-mediated gene silencing. Cell.
132:9–14. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li X, Kong D, Chen H, Liu S, Hu H, Wu T,
Wang J, Chen W, Ning Y, Li Y and Lu Z: miR-155 acts as an
anti-inflammatory factor in atherosclerosis-associated foam cell
formation by repressing calcium-regulated heat stable protein 1.
Sci Rep. 6:217892016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu Z, Han Y, Liu J, Jiang F, Hu H, Wang Y,
Liu Q, Gong Y and Li X: miR-135b-5p and miR-499a-3p promote cell
proliferation and migration in atherosclerosis by directly
targeting MEF2C. Sci Rep. 5:122762015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Qin W, Zhang L, Wu X, Du N, Hu Y,
Li X, Shen N, Xiao D, Zhang H, et al: MicroRNA-26a prevents
endothelial cell apoptosis by directly targeting TRPC6 in the
setting of atherosclerosis. Sci Rep. 5:94012015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ouimet M, Ediriweera HN, Gundra UM, Sheedy
FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C,
Fullerton MD, Cecchini K, et al: MicroRNA-33-dependent regulation
of macrophage metabolism directs immune cell polarization in
atherosclerosis. J Clin Invest. 125:4334–4348. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen T, Huang Z, Wang L, Wang Y, Wu F,
Meng S and Wang C: MicroRNA-125a-5p partly regulates the
inflammatory response, lipid uptake, and ORP9 expression in
oxLDL-stimulated monocyte/macrophages. Cardiovasc Res. 83:131–139.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu J, Chen T, Yang L, Li Z, Wong MM,
Zheng X, Pan X, Zhang L and Yan H: Regulation of microRNA-155 in
atherosclerotic inflammatory responses by targeting MAP3K10. PLoS
One. 7:e465512012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Daigneault M, Preston JA, Marriott HM,
Whyte MK and Dockrell DH: The identifcation of markers of
macrophage differentiation in PMA-stimulated THP-1 cells and
monocyte-derived macrophages. PLoS One. 5:e86682010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bao Y, Wang L, Xu Y, Yang Y, Wang L, Si S,
Cho S and Hong B: Salvianolic acid B inhibits macrophage uptake of
modifed low density lipoprotein (mLDL) in a scavenger receptor
CD36-dependent manner. Atherosclerosis. 223:152–159. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu GF, Yang LX, Guo RW, Liu H, Shi YK,
Wang H, Ye JS, Yang ZH and Liang X: miR-155 inhibits oxidized
low-density lipoprotein-induced apoptosis of RAW264.7 cells. Mol
Cell Biochem. 382:253–261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weber C and Noels H: Atherosclerosis:
Current pathogenesis and therapeutic options. Nat Med.
17:1410–1422. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zernecke A, Bidzhekov K, Noels H,
Shagdarsuren E, Gan L, Denecke B, Hristov M, Köppel T, Jahantigh
MN, Lutgens E, et al: Delivery of microRNA-126 by apoptotic bodies
induces CXCL12-dependent vascular protection. Sci Signal.
2:ra812009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schober A, Nazari-Jahantigh M, Wei Y,
Bidzhekov K, Gremse F, Grommes J, Megens RT, Heyll K, Noels H,
Hristov M, et al: MicroRNA-126-5p promotes endothelial
proliferation and limits atherosclerosis by suppressing Dlk1. Nat
Med. 20:368–376. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tabet F, Vickers KC, Torres Cuesta LF,
Wiese CB, Shoucri BM, Lambert G, Catherinet C, Prado-Lourenco L,
Levin MG, Thacker S, et al: HDL-transferred microRNA-223 regulates
ICAM-1 expression in endothelial cells. Nat Commun. 5:32922014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Liu D, Chen X, Li J, Li L, Bian
Z, Sun F, Lu J, Yin Y, Cai X, et al: Secreted monocytic miR-150
enhances targeted endothelial cell migration. Mol Cell. 39:133–144.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fang H, Lin J, Wang L, Xie P, Wang X, Fu
J, Ai W, Chen S, Chen F, Zhang F, et al: Kruppel-like factor 2
regulates dendritic cell activation in patients with acute coronary
syndrome. Cell Physiol Biochem. 32:931–941. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meng Z, Yan C, Deng Q, Dong X, Duan ZM,
Gao DF and Niu XL: Oxidized low-density lipoprotein induces
inflammatory responses in cultured human mast cells via Toll-like
receptor 4. Cell Physiol Biochem. 31:842–853. 2013. View Article : Google Scholar : PubMed/NCBI
|