Introduction
Alzheimer's disease (AD), first described and termed
by the German psychiatrist and pathologist Alois Alzheimer in 1906,
is a chronic neurodegenerative disease and a form of dementia
(1,2). There are ~47 million patients with
dementia worldwide with 9.9 million new cases every year, in which
60–80% of dementia cases are AD (2).
In AD, the patient's mental and physical condition declines,
including problems with language, orientation, memory and
motivation, ultimately leading to mortality (2,3). Senile
plaques are one of the most important pathological features of AD.
AD is highly associated with senile plaques due to the accumulation
of amyloid β (Aβ) protein (4).
Besides increasing age and certain genetic risks, AD is associated
with inflammatory cytokines, vascular diseases and cholesterol
levels (5–7). Currently, a number of medications or
supplements can efficiently attenuate the progression or decrease
the risk of developing AD (8).
p38 mitogen-activated protein kinase (MAPK) is
responsive to stress stimuli, including inflammation, cytokines,
radiation and shock, and is involved in cell proliferation,
differentiation, apoptosis and autophagy. Given that AD is a
neurodegenerative disease associated with inflammation and
cytokines (5,9), investigating the role of p38 MAPK will
aid in the understanding of the underlying mechanisms of AD.
Cytochromes P450 (CYPs; max absorption, 450 nm) is a superfamily of
the terminal oxidase enzymes that are present in electron transfer
chains, in which electrons are transferred from NADPH to CYPs via
cytochrome P450 reductase (CPR). CYPs are involved in the majority
of chemical metabolism (deactivation or activation) that occurs in
the body. Certain chemicals can change the biosynthesis or activity
of CYPs, which then affects the metabolism and clearance of other
compounds (10,11). Although certain CYPs have been
reported to be abnormally expressed in AD (12–14),
reports on CPR are limited.
In the present study, in vivo and in
vitro studies were performed to investigate the role of p38
MAPK and CPR in AD. The results of the present study may aid in the
understanding of the mechanisms of and potential treatments for
AD.
Materials and methods
Control mice and mouse models of
AD
APPswe/PSEN1dE9 (AD) male mice (weighing 24±3 g at 9
months old), originally generated in Jackson Laboratory (Bar
Harbor, ME USA), were purchased from the Model Animal Research
Center of Nanjing University (Nanjing, China); age- and sex-matched
wild-type (WT) C57BL/6J male mice (weighing 22±2 g) were also were
purchased from Model Animal Research Center of Nanjing University
and used as negative controls. A total of 36 mice, 21 AD mice and
15 WT mice were used. Among which, 9 AD and 3 WT mice were treated
with SB203580 (named as AD + SB and WT + SB separately). The mice
were raised in an environment with a temperature of 24±2°C,
humidity of 55±15%, and access to a 12-h light/dark cycle. Mice
were given ad libitum access to food and water. Experiments
and surgeries on the mice were performed according to the
Institutional Animal Care and Use Committee (IACUC) guidelines. The
animal experiments were approved by and performed in the Animal
Research Center of Suzhou Vocational Health College (Suzhou,
China).
Senile plaque staining and
determination of contextual memory
A total of 12 mice were selected in the senile
plaque study. A total of 3 WT mice and 3 AD mice were treated with
SB203580 (10 mg/kg; intraperitoneal injection; Sigma-Aldrich; Merck
KGaA; Darmstadt, Germany) every 3 days for 30 days starting at the
9-month time point, experiments were then performed at the 10-month
time point. There was a total of four groups of 10-month-old mice,
as follows: WT, AD, WT + SB and AD + SB (3 mice in each group).
Brain damage in the AD mice was tested using Thioflavin S
(Sigma-Aldrich; Merck KGaA) staining. Mice were euthanized by
exposing them to CO2 following the IACUC guidelines, the
brains were then dissected and embedded in optimum cutting
temperature compound, and frozen on dry ice. The coronal sections
of the mouse brains were cut into 10-µm-thick sections, placed onto
slides, and then fixed in 4% paraformaldehyde (PFA) at room
temperature for 20 min. The slides were placed in 1% Thioflavin S
solution at room temperature for 20 min and then differentiated
twice in 70% fresh alcohol for 7 min. Images were captured using a
confocal microscope (magnification, ×100) and analyzed by
calculating the relative area of staining using ImageJ software
(National Institutes of Health, Bethesda, MD, USA).
The contextual memory of the mice was examined using
a fear-conditioning assay modified from a previous study (15). A total of 6 WT mice, 6 AD mice and 6
AD + SB mice were used for the contextual memory study. Freezing
tests were performed in four chambers (17.8×19.1×38.1 cm). The
session began with the house light on. Firstly, mice were exposed
to 5 conditioned stimuli (30 sec, 85 dB, white noise), followed by
a 30-sec trace period and treatment with the unconditioned stimulus
(2 sec, 0.285-mA foot-shock) paired with a 5-sec tone in the
chambers on the first day. On the second day, mice were placed in
the chambers to test for freezing for a total of 5 min. Freezing
time in the absence of foot-shock and tone in the first 2.5 min and
with the tone in the later 2.5 min were recorded. Relative memory
deficit was calculated as the percentage of freezing condition
(absence of all non-respiratory movements) as follows: Freezing
time/Total test time ×100%.
Detection of p-p38 MAPK and CPR in
mouse cortexes by western blot analysis
phospho (p)-p38 MAPK and CPR were detected in the
cortexes of the brains of 3 WT mice and 3 AD mice. Microsomes were
obtained and isolated from the cortexes; these subcellular
fractions were used to detect the expression of CPR. The cortex was
homogenized on ice in 10 mM PBS (pH, 7.7) containing 250 mM
sucrose, 1 mM EDTA and 0.5 mM phenylmethylsulfonyl fluoride for 15
sec, then centrifuged for 20 min at 10,000 × g at 4°C. The
supernatant was collected and centrifuged for 1 h at 40,000 × g at
4°C, then the supernatant was decanted. The pellet was resuspended
in 0.1 M PBS containing 1 mM EDTA, 1 mM dithiothreitol, 30%
glycerol and protease inhibitors (pH, 7.25). Protein was also
extracted from the mouse cortexes and homogenized in protein
extraction buffer containing 0.5 mM phenylmethylsulfonyl fluoride,
80 µg/ml DL-dithiothreitol. Protein was boiled for 5 min, cooled on
ice for 1 min and then centrifuged for 1 min at 10,000 × g at 4°C.
Both microsome and protein were quantified using BCA Protein Assay
(Thermo Scientific, Rockford, IL, USA). A total of 10 µg microsome
or 25 µg protein were injected into each lane then separated via
SDS-PAGE on a 12.5% gel and transferred onto a polyvinylidene
difluoride membrane. The membranes were blocked in 5% fat-free milk
for 1 h at room temperature, then incubated overnight at 4°C with a
primary rabbit polyclonal IgG antibodies directed against p-p38
MAPK (Thr180/Tyr182; cat no: 9211; 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA) or CPR (cat. no. ab13513;
1:1,000; Abcam, Cambridge, UK). Subsequently, secondary anti-rabbit
goat IgG antibodies (cat. no. A16110; 1:4,000; Thermo Fisher
Scientific, Inc., Carlsbad, CA, USA) were applied for 2 h at room
temperature. GAPDH (cat. no. G9545; 1:2,000; Sigma-Aldrich, Merck
KGaA) was used as internal control for protein detection.
Detection of p-p38 MAPK and CPR in
SH-SY5Y cells
The human neuroblastoma cell line, SH-SY5Y, was
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). The cells were cultured in a 1:1
mixture of DMEM and Ham's Nutrient Mixture F12 medium (both
HyClone™; GE Healthcare, Logan, UT, USA) supplemented
with 10% fetal bovine serum (Shanghai ExCell Biology, Inc.,
Shanghai, China), 100 U/ml penicillin and 0.1 mg/ml streptomycin
(HyClone; GE Healthcare). The sequence of the Aβ1–42
peptide (molecular weight, 4514.08 kDa; Sangon Biotech Co., Ltd.,
Shanghai, China) was DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA.
Aβ1–42 was dissolved in dimethyl sulfoxide, giving a
concentration of 1 mM; 100 µl of this solution and 900 µl PBS was
then mixed to generate a 100 µM Aβ1–42 solution. The
product was incubated at 37°C for 7 days prior to usage. SH-SY5Y
cells were treated with 10 µM Aβ1–42 for 48 h prior to
the detection of p-p38 MAPK in the cell lysate (previously
subjected ice-cold cell lysis buffer before the pellet was
resuspended, incubated on ice for 10 min, centrifuged at 20,817 × g
for 15 min at 4°C and the supernatant was collected) and CPR in
microsomes by western blotting as described above.
Cell viability and apoptosis
assays
MTT assays were performed to detect the viability of
SH-SY5Y cells, which were seeded at a density of 3,000 cells/well
in 96-well plates and cultured with 10 µM Aβ1–42 for 72
h. The expression of p38 MAPK and CPR was inhibited using the
following inhibitors: 3 µM SB203580 (SB) (Sigma-Aldrich; Merck
KGaA) and 80 µM tannic acid (TA) (Sigma-Aldrich; Merck KGaA),
respectively. For the proliferation assay, 20 µl of 5 mg/ml MTT
(Beyotime Institute of Biotechnology, Haimen, China) was added to
each well and the plates were incubated at 37°C for 4 h. The medium
was decanted and the cells were incubated with 150 µl formazan
solution at 37°C for 15 min. The absorbance of the wells at 590 nm
in four groups (WT, 10 µM Aβ1–42, 10 µM
Aβ1–42 + SB203580 and 10 µM Aβ1–42 + TA) was
then measured. Cell viability in WT group was set as 100%, data in
other groups were calculated by comparison.
Staining with 4′,6-diamidino-2-phenylindole (DAPI;
Beyotime Institute of Biotechnology) was performed to detect
apoptotic cells. Subsequent to fixing with 4% PFA for 20 min and 1X
PBS/0.5% Triton X-100 for 10 min on ice, the cells were stained
with DAPI solution at room temperature for 15 min. Images were
captured using a fluorescence microscope (Eclipse Ti-S; Nikon
Corporation, Tokyo, Japan) at a magnification of ×200. Caspase-3
activity was detected using Caspase 3 Activity Assay kit (Beyotime
Biotechnology), according to the manufacturer's protocol. A total
of 10 µl of 2 mM Ac-DEVD-pNA, a caspase-3 substrate that hydrolyzes
to pNA, was added to the SH-SY5Y cell culture, which was then
incubated for 2 h at 37°C. The absorbance of pNA at 405 nm was
subsequently measured. The activity of caspase-3 was normalized to
the total concentration of protein in the lysates and expressed as
the relative value. The negative control (NC) group consisted of
untreated cells was set as 1.
Statistical analysis
Data are presented as the mean ± standard deviation,
and were analyzed using SPSS software (version 16.0; SPSS, Inc.,
Chicago, IL, USA). Results were evaluated using the Student's
t-test for two groups or one-way analysis of variance followed by a
post hoc Dunnett's test for multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
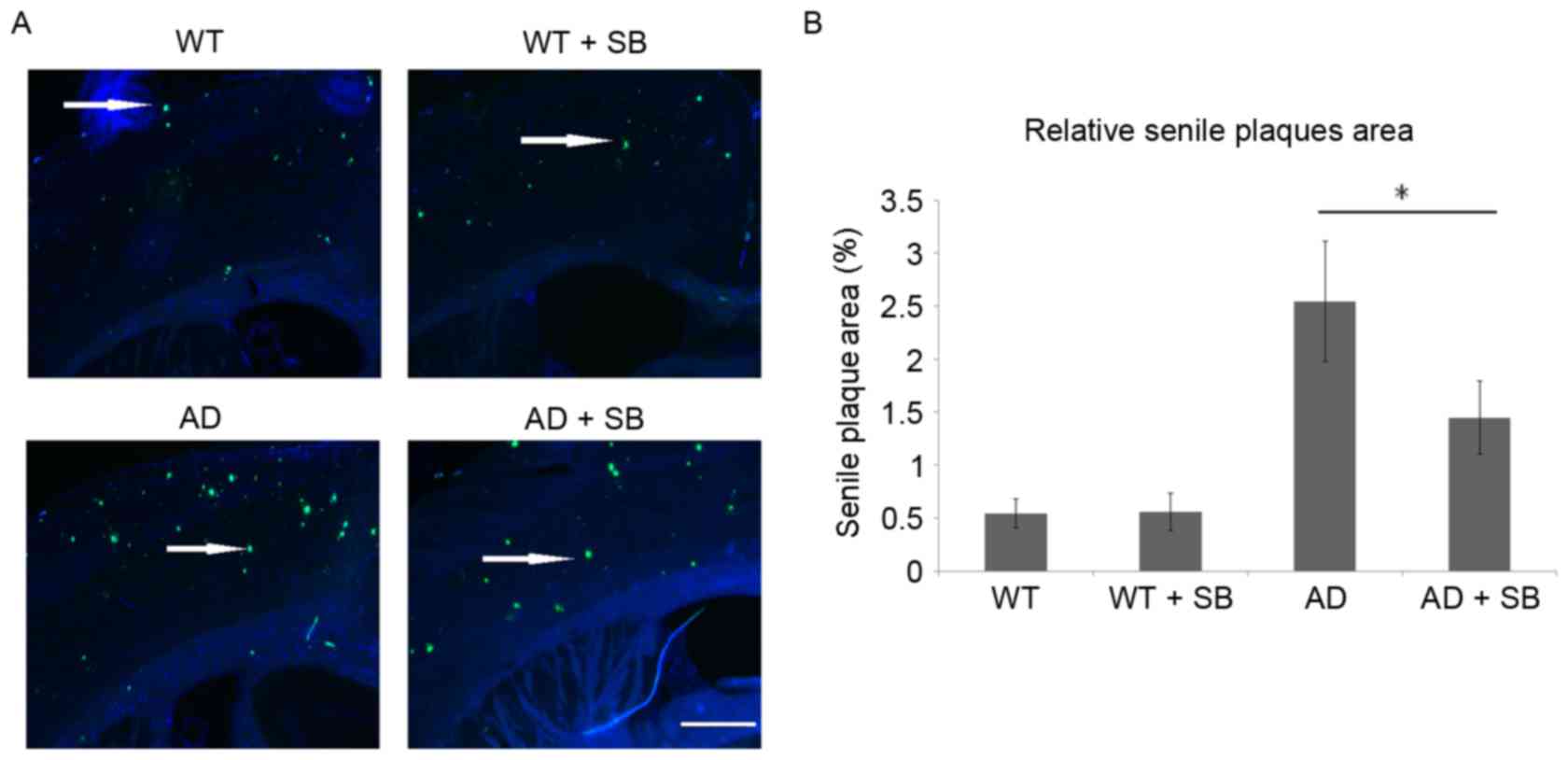
Inhibition of p38 MAPK decreases the
relative senile plaque area in the brains of mice with AD
Senile plaques in mouse cortexes on the hippocampal
side were detected using Thioflavin S staining and the relative
area of senile plaques in the whole section was evaluated (Fig. 1). A number of senile plaques were
detected in the WT and WT + SB groups with no obvious differences.
The area of senile plaques increased markedly in the AD group
compared with the WT group, indicating senile plaque formation and
accumulation. Inhibition of p38 MAPK with SB203580 significantly
decreased the area of senile plaques in these AD mice (P<0.05;
Fig. 1B).
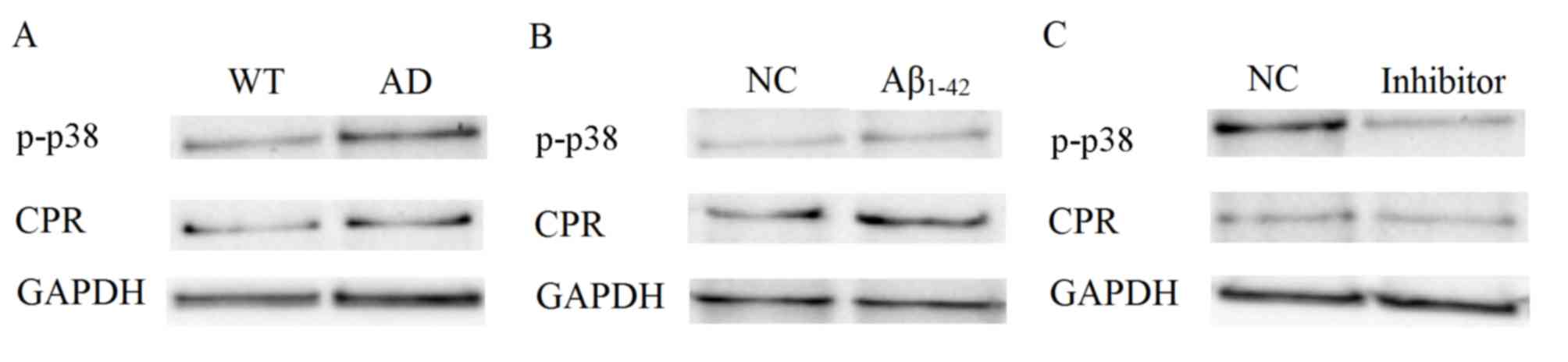
Expression of p-p38 MAPK and CPR is
decreased in AD mice and Aβ1–42-treated SH-SY5Y
cells
p-p38 MAPK and CPR in mouse brain cortexes and cells
were detected by western blot analyses. p-p38 and CPR were notably
upregulated in the mice brain cortex in the AD group compared with
the WT group (Fig. 2A). p-p38 and
CPR were also upregulated in SY-SH5Y cells treated with 10 µM
Aβ1–42 compared with untreated cells (Fig. 2B). The expression levels of p-p38 and
CPR decreased following the treatment with their respective
inhibitors (Fig. 2C). These results
indicate that CPR and p38 MAPK serve complex roles in AD.
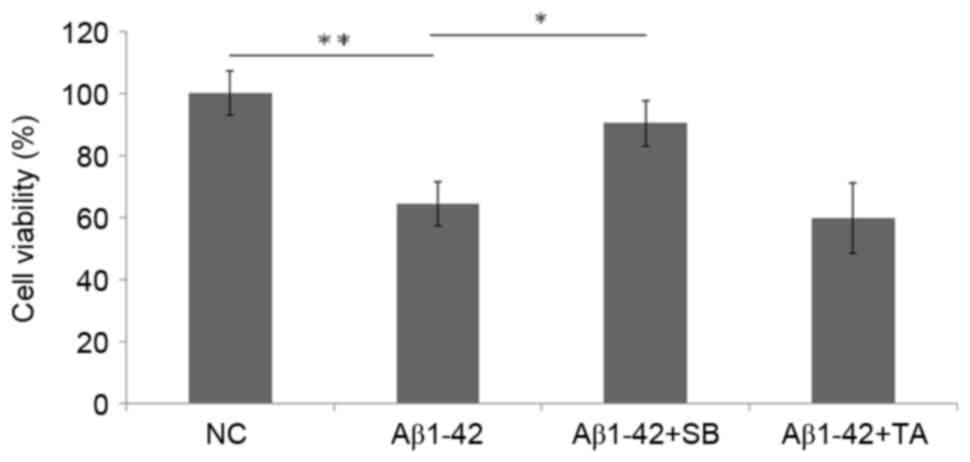
Neuroblastoma cell viability is
inhibited by Aβ1–42, but this is attenuated by p38 MAPK
inhibition
The viability of SH-SY5Y cells was significantly
inhibited due to neural toxicity following 10 µM Aβ1–42
treatment (P<0.01 vs. the NC group; Fig. 3). Following the inhibition of p38
MAPK with SB203580, the viability of SH-SY5Y cells treated with
Aβ1–42 significantly increased compared with the 10 µM
Aβ1–42 alone group (P<0.05; Fig. 3). No significant differences were
identified in the Aβ1–42 + TA group compared with the
Aβ1–42 group. Thus, no protection was identified when
CPR was inhibited.
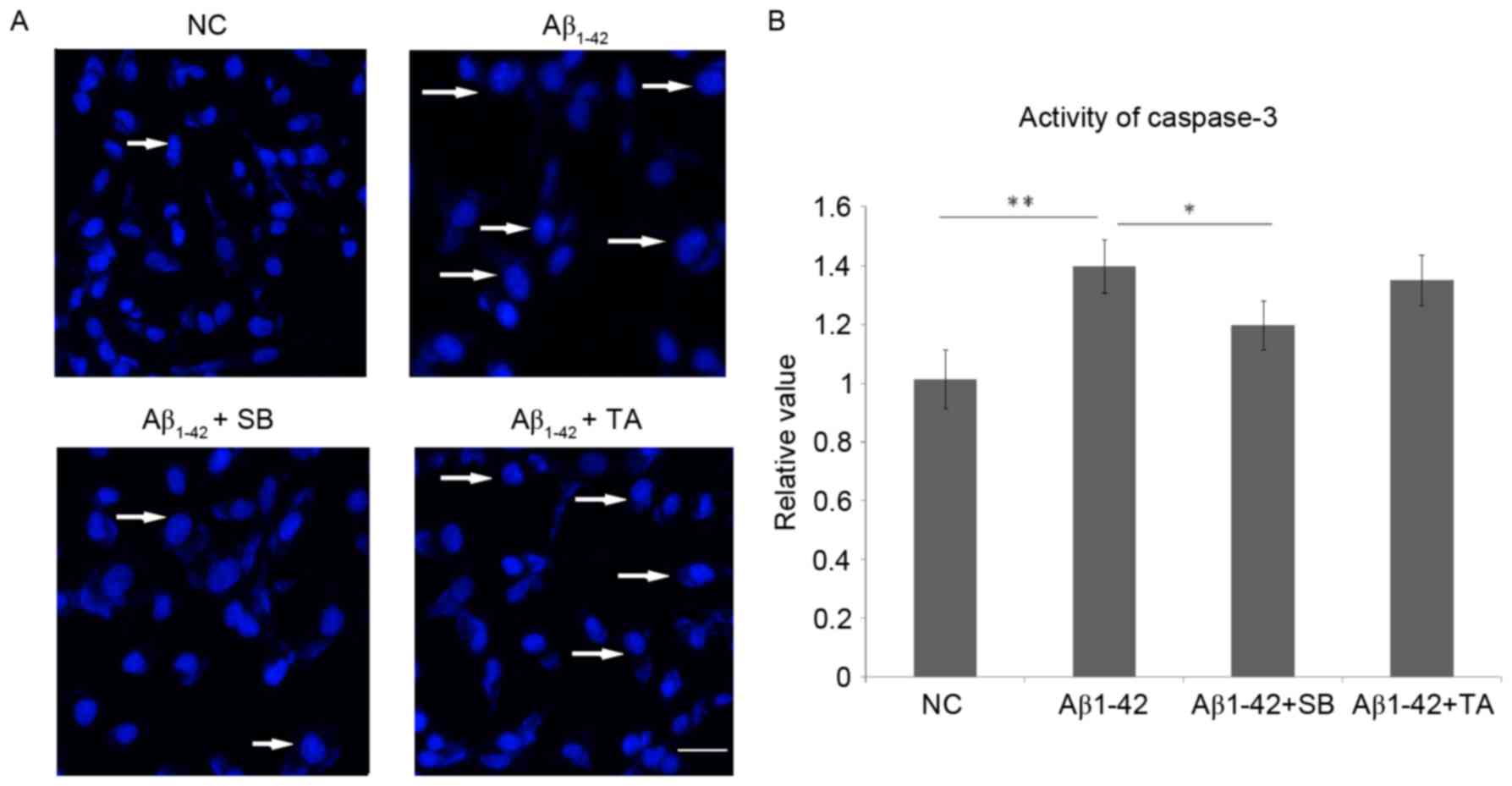
Neuroblastoma cell
Aβ1–42-induced apoptosis is attenuated by p38 MAPK, but
not CPR, inhibition
DAPI staining indicated that inhibition of p38 MAPK
could protect SH-SY5Y cells from apoptosis caused by the neural
toxicity of Aβ1–42 (Fig.
4). Inhibition of CPR yielded no differences in the level of
apoptosis (Fig. 4). The activity of
caspase-3 was significantly increased following treatment with 10
µM Aβ1–42 compared with the untreated cells (P<0.01;
Fig. 4B). However, caspase-3
activity was significantly decreased in Aβ1–42-treated
cells after SB203580 treatment compared with cells treated with
Aβ1–42 alone (P<0.05; Fig
4A), while no changes were identified in the cells treated with
Aβ1–42 and TA. These results suggest that the inhibition
of p38 MAPK protects SH-SY5Y cells from apoptosis.
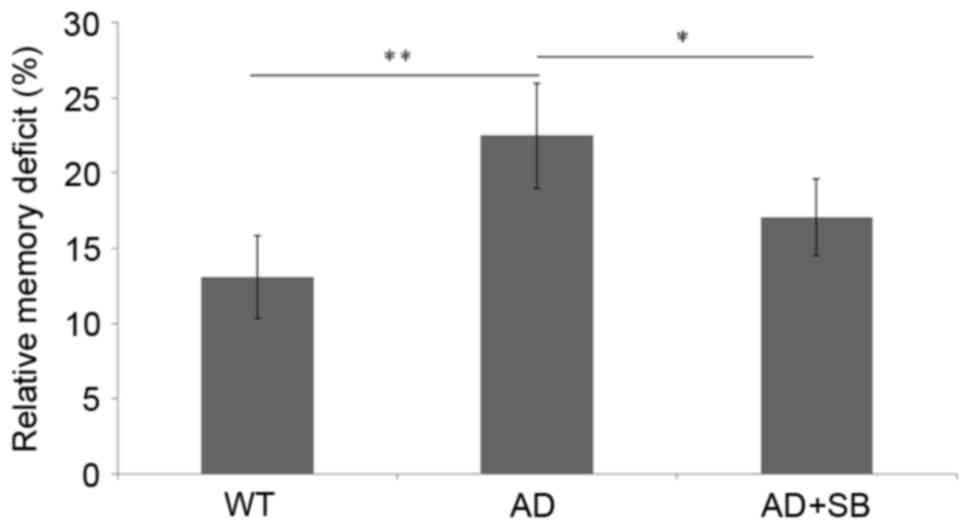
p38 MAPK inhibition promotes
contextual memory improvement
No significant differences were identified in
relative memory deficit between the WT and SB203580 treated WT
mice, suggesting that SB203580 is not toxic to normal cognitive
function (data not shown). However, the AD mice demonstrated
significant memory deficiencies compared with the WT mice
(P<0.01; Fig. 5), indicating
impaired contextual memory. Following the inhibition of p38 MAPK,
the contextual memory of SB-treated AD mice significantly improved
compared with untreated AD mice (P<0.05), although was still
deficient compared with the WT mice (Fig. 5). These results suggest that the
deficiency in contextual memory identified in AD mice is due to
neural toxicity and could be improved by inhibition of p38
MAPK.
Discussion
AD is a chronic neurodegenerative disease for which
there are no effective drug treatments. At present, the morbidity
of AD is increasing worldwide. However, the mechanism of the
development of AD remains unclear. The present study aimed to
elucidate the role of p38 MAPK and CPR in AD, in animals and at the
cellular level. The AD mouse model ASPswe/PSEN1dE9 encoding a
chimeric Aβ (A4) precursor protein (APPswe) and mutation of human
presenilin 1 was generated in the Jackson Laboratory. Thioflavin S
staining was then applied to detect senile plaques. As neuritic
deposits consisting of Aβ peptides, such as Aβ1–42, in
the cortex of the brain, senile plaques are variable in shape, area
and size, and are associated with degenerative neural structures,
and an abundance of microglia and astrocytes (16). Senile plaques, in which abnormal
neurites are composed primarily of paired helical filaments and
neurofibrillary tangles, are one of the most important
characteristic features of AD. Aβ1–42 tends to aggregate
into senile plaques and is neurotoxic (16–18). The
present study revealed that senile plaques were seldom identified
in healthy mice in the WT group. The number of senile plaques was
greater in the AD group, indicating that AD mice were successfully
generated.
Using this AD mouse model, the expression of p38
MAPK was examined. p38 MAPK, also called cytokinin-specific binding
protein, is the mammalian orthologue of the yeast protein, Hog1p
MAP kinase. p38 MAPK is involved in a signaling cascade controlling
cellular responses, similar to the stress-activated protein
kinase/c-Jun N-terminal kinase signaling pathway (19). In the present study, p-p38 MAPK was
demonstrated to be significantly upregulated in the brain cortex of
mice in the AD group. p38 MAPK can be activated by a variety of
cellular stressors. In a previous study by our group, p38 MAPK
could be activated by inflammatory cytokines, lipopolysaccharides
and growth factors in mice brains (unpublished). This previous
study indicated that the inflammation was induced and p38 MAPK was
involved in AD. Whether the inflammation in the brain was primarily
induced through the p38 MAPK signaling pathway remains unclear and
requires further study. The conclusion from the previous study by
our group was investigated in the present study by experiments on
the human SH-SY5Y neuroblastoma cell line, whose original cell
line, SK-N-SH, was isolated from a bone marrow biopsy obtained from
a 4-year-old female with neuroblastoma. SH-SY5Y cells are widely
used for in vitro neuronal function and differentiation
studies. Given that AD is a human neurodegenerative disease, these
human cells were selected instead of mouse neurons.
Aβ peptides are formed through sequential cleavage
of the amyloid precursor protein. Aβ1–42 is associated
with AD; thus, Aβ1–42 was used as a neural toxin to
treat the SH-SY5Y cells. The present study identified that p-p38
MAPK was significantly upregulated in SH-SY5Y cells treated with
Aβ1–42, indicating that p38 MAPK is a potential target
for the treatment of AD.
Another important enzyme in AD, CPR, was
investigated in the current study. CPR, a microsomal flavoprotein
and redox partner of CYPs, is required for all monooxygenase
reactions catalyzed by microsomal CYPs, shuttling electrons from
NADPH into the iron of the prosthetic heme group of CYPs through
FAD and FMN coenzymes (20). CPR is
located in the inner membrane of the mitochondria and in the
endoplasmic reticulum of cells, and is present in the majority of
tissues of the body, such as the brain, metabolizing thousands of
endogenous and exogenous chemicals. CPR is one of the main
drug-metabolizing enzymes in the human body. Given that the aim of
the present study was to evaluate the potential to target p38 MAPK
in the treatment of AD, the role of CPR was also studied. A
previous study from our group indicated that CPR was highly
associated with astrocytosis (21).
Along with p38 MAPK, CPR was also significantly upregulated in the
cortex of mice in the AD group. However, the inhibition of CPR in
SH-SY5Y cells indicated no significant difference compared with the
cells treated with Aβ1–42 alone. Thus, the role of CPR
in the regulation of AD appears to be complex.
Given that p-p38 MAPK and CPR levels were increased
in the cortexes of the brains of mice in the AD group and SH-SY5Y
cells treated with Aβ1–42, p38 MAPK and CPR were
downregulated by inhibitor treatment in order to investigate their
effect on cell viability and apoptosis. MTT assays were performed
to analyze the viability of SH-SY5Y cells. Due to its neural
toxicity, Aβ1–42 significantly inhibited cell viability.
Inhibition of p38 MAPK promoted the viability of SH-SY5Y cells
treated with Aβ1–42. DAPI staining also confirmed that
inhibition of p38 MAPK protected SH-SY5Y cells from apoptosis
caused by the neural toxicity of Aβ1–42. Inhibition of
CPR demonstrated no significant benefits to cellular viability or
apoptosis. To confirm this effect, the activity of caspase-3, which
is activated in apoptotic cells and serves as a key indicator for
apoptosis, was analyzed. Caspase-3 was significantly increased in
SH-SY5Y cells that were treated with Aβ1–42 and
significantly decreased following the inhibition of p38 MAPK.
However, CPR inhibition demonstrated no significant effect on
apoptosis.
Based on the in vitro experiments performed
in the present study, in vivo experiments were conducted. A
fear-conditioning assay was performed to determine the contextual
memory of the mice, since contextual memory deficiency is one of
the main symptoms of AD (22). Due
to neural toxicity, deficiencies in contextual memory were
identified in AD mice. Notably, inhibition of p38 MAPK was revealed
to improve contextual memory in AD mice.
In conclusion, the results of the present study
indicate that p38 MAPK is overactive in AD. CPR was also involved
in this process; however, its activity in the regulation of AD was
more complex than p38 MAPK. Due to neural toxicity, the number of
senile plaques increased and deficiencies in contextual memory were
identified in the AD mice. These symptoms were reduced by p38 MAPK
inhibition. Inhibition of p38 MAPK was also demonstrated to protect
Aβ1–42-treated SH-SY5Y cells from apoptosis and promote
their viability. These findings provide novel insights into the
pathogenesis of AD and highlight potential targets for AD
treatment.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Jiangsu Province (grant no. BK20130219), the
National Nature Science Foundation of China (grant no. 81401045)
and the Qing Lan Project of Jiangsu Province.
References
|
1
|
Shampo MA, Kyle RA and Steensma DP: Alois
Alzheimer-Alzheimer disease. Mayo Clin Proc. 88:pp. e1552013;
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alzheimer's Association. 2017 Alzheimer's
disease facts and figures. Alzheimers Dement. 13:325–373. 2017.
View Article : Google Scholar
|
|
3
|
Nithianantharajah J and Hannan AJ: The
neurobiology of brain and cognitive reserve: Mental and physical
activity as modulators of brain disorders. Prog Neurobiol.
89:369–382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arbor SC, LaFontaine M and Cumbay M:
Amyloid-beta Alzheimer targets-protein processing, lipid rafts, and
amyloid-beta pores. Yale J Biol Med. 89:5–21. 2016.PubMed/NCBI
|
|
5
|
Su F, Bai F and Zhang Z: Inflammatory
cytokines and Alzheimer's disease: A review from the perspective of
genetic polymorphisms. Neurosci Bull. 32:469–480. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lénárt N, Brough D and Dénes Á:
Inflammasomes link vascular disease with neuroinflammation and
brain disorders. J Cereb Blood Flow Metab. 36:1668–1685. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hall JR, Wiechmann AR, Johnson LA, Edwards
M, Barber RC, Cunningham R, Singh M and O'Bryant SE: Total
cholesterol and neuropsychiatric symptoms in Alzheimer's disease:
The impact of total cholesterol level and gender. Dement Geriatr
Cogn Disord. 38:300–309. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rea R, Carotenuto A, Fasanaro AM, Traini E
and Amenta F: Apathy in Alzheimer's disease: Any effective
treatment? Scientific World Journal 2014. 4213852014.
|
|
9
|
Zheng C, Zhou XW and Wang JZ: The dual
roles of cytokines in Alzheimer's disease: Update on interleukins,
TNF-α, TGF-β and IFN-γ. Transl Neurodegener. 5:72016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tompkins LM and Wallace AD: Mechanisms of
cytochrome P450 induction. J Biochem Mol Toxicol. 21:176–181. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Souslova T, Marple TC, Spiekerman AM and
Mohammad AA: Personalized medicine in Alzheimer's disease and
depression. Contemp Clin Trials. 36:616–623. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu J, Wan L, Zhong Y, Yu Q, Han Y, Chen P,
Wang B, Li W, Miao Y and Guo C: Stereoselective metabolism of
donepezil and steady-state plasma concentrations of S-donepezil
based on CYP2D6 polymorphisms in the therapeutic responses of Han
Chinese patients with Alzheimer's disease. J Pharmacol Sci.
129:188–195. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan H, Kong Y, He B, Huang M, Li J, Zheng
J, Liang L, Bi J, Zhao S and Shi L: CYP2J2 rs890293 polymorphism is
associated with susceptibility to Alzheimer's disease in the
Chinese Han population. Neurosci Lett. 593:56–60. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mast N, Li Y, Linger M, Clark M, Wiseman J
and Pikuleva IA: Pharmacologic stimulation of cytochrome P450 46A1
and cerebral cholesterol turnover in mice. J Biol Chem.
289:3529–3538. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bolivar VJ, Manley K and Messer A:
Exploratory activity and fear conditioning abnormalities develop
early in R6/2 Huntington's disease transgenic mice. Behav Neurosci.
117:1233–1242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mohri I, Kadoyama K, Kanekiyo T, Sato Y,
Kagitani-Shimono K, Saito Y, Suzuki K, Kudo T, Takeda M, Urade Y,
et al: Hematopoietic prostaglandin D synthase and DP1 receptor are
selectively upregulated in microglia and astrocytes within senile
plaques from human patients and in a mouse model of Alzheimer
disease. J Neuropathol Exp Neurol. 66:469–480. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nathalie P and Jean-Noël O: Processing of
amyloid precursor protein and amyloid peptide neurotoxicity. Curr
Alzheimer Res. 5:92–99. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iqbal K and Grundke-Iqbal I: Alzheimer
neurofibrillary degeneration: Significance, etiopathogenesis,
therapeutics and prevention. J Cell Mol Med. 12:38–55. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kashimata M, Sayeed S, Ka A, Onetti-Muda
A, Sakagami H, Faraggiana T and Gresik EW: The ERK-1/2 signaling
pathway is involved in the stimulation of branching morphogenesis
of fetal mouse submandibular glands by EGF. Dev Biol. 220:183–196.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Laursen T, Jensen K and Møller BL:
Conformational changes of the NADPH-dependent cytochrome P450
reductase in the course of electron transfer to cytochromes P450.
Biochim Biophys Acta. 1814:132–138. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao Y, Liu S, Wang Y, Yuan W, Ding X,
Cheng T, Shen Q and Gu J: Suppression of cytochrome P450 reductase
expression promotes astrocytosis in subventricular zone of adult
mice. Neurosci Lett. 548:84–89. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Webster SJ, Bachstetter AD, Nelson PT,
Schmitt FA and Van Eldik LJ: Using mice to model Alzheimer's
dementia: An overview of the clinical disease and the preclinical
behavioral changes in 10 mouse models. Front Genet. 5:882014.
View Article : Google Scholar : PubMed/NCBI
|