Introduction
Atrial fibrillation (AF) is the most common type of
arrhythmia and is associated with increased cardiovascular
morbidity and mortality (1). Current
drug treatments of AF have moderate efficacy and numerous
limitations, including proarrhythmia and bleeding complications in
particular (2). An enhanced
understanding of the molecular mechanisms of AF is required to
develop novel interventions for AF.
Although the pathogenesis of AF has remained to be
fully elucidated, focal ectopic activity and re-entry are thought
to be the two major determinants of AF initiation and maintenance
independent of the underlying causes (2). The decelerated conduction and shortened
refractoriness contribute to the formation of re-entry (3). The gap junction, which connects the
cytoplasm of adjacent cells, is a key regulator of conduction in
the heart (4). Gap junctions are
clustered channels, consisting of two hemichannels, each of which
is formed by six connexins (Cxs) (5). Cx40 is one of the principal atrial gap
junction proteins (6). Igarashi
et al (7) have demonstrated
that gene therapy with adenovirus expressing Cx40 or Cx43 preserves
atrial conduction and prevents AF in a swine model. This indicated
that interventions targeting Cx40 may be promising for AF therapy.
However, the specific regulatory mechanisms of Cx40, particularly
at the post-transcriptional level, remain to be fully elucidated in
human AF.
microRNAs (miRs) are a class of small, non-coding
RNAs of ~22 nucleotides in length, which negatively regulate target
gene expression at the post-transcriptional level (8). They generally bind to a complementary
sequence in the 3′-untranslated regions (3′UTRs) of their target
mRNAs, which results in degradation of the mRNA or inhibition of
its translation (8). In recent
years, a number of studies have demonstrated that dysregulation of
certain miRs is associated with AF-maintaining substrates by
promoting atrial remolding, including electrical and structural
remolding (9,10). Previous studies have indicated that
miR-208 may be involved in the pathogenesis of AF (11,12), but
at present, the exact role of miR-208 in human AF is poorly
understood. Of note, preliminary bioinformatics analyses revealed
that CJA5 encoding Cx40 is a potential target gene of miR-208a-3p.
Therefore, the present study investigated whether miR-208a-3p was
responsible for AF by targeting Cx40 directly.
Materials and methods
Collection of human atrial
samples
A total of 37 patients with valvar heart disease
undergoing cardiac surgery at the First Affiliated Hospital,
Guangxi Medical University between March 2015 and August 2015 were
divided into a chronic AF group [19 cases with long-standing,
persistent AF (13); 9 males, 10
females; average age, 51.53±9.81 years] and a sinus rhythm (SR)
group (8 males, 10 females; average age, 47.78±10.10 years;
Table I). The diagnosis of AF was
reached by evaluating medical records and 12-lead electrocardiogram
findings. Those who had hypertension, diabetes mellitus, coronary
heart disease, infective endocarditis, active rheumatism, pulmonary
disease, hyperthyroidism or autoimmune disease were excluded from
this study. The protocol was in accordance with the Helsinki
Declaration and was approved by the Human Ethics Committee of the
First Affiliated Hospital of Guangxi Medical University (Guangxi,
China). All patients enrolled in this study provided written
informed consent.
 | Table I.Characteristics of patients with SR or
AF. |
Table I.
Characteristics of patients with SR or
AF.
| Characteristics | SR group (n=18) | AF group (n=19) | P-values |
|---|
| Age (years) | 47.78±10.10 | 51.53±6.14 | 0.179 |
| Sex (male) | 8 (44.4) | 9 (47.4) | 0.746 |
| Cardiac disease
type |
|
|
|
| MS | 0 | 2 (10.5) | – |
| MI | 3 (16.7) | 1 (5.3) | 0.604 |
|
MS+MI | 1 (5.6) | 2 (10.5) | 0.604 |
| AI | 1 (5.6) | 0 | – |
|
CVL | 13 (72.2) | 14 (73.7) | 0.714 |
| Left atrial
thrombus | 1 (5.6) | 6
(31.6)a | 0.090 |
| LAD (mm) | 46.06±7.57 |
65.16±9.81a | P<0.001 |
| LVEF (mm) | 62.75±9.69 | 63.32±7.54 | 0.844 |
| NYHA class |
|
|
|
| II | 12 (66.7) | 7 (36.8) | 0.194 |
|
III | 5 (27.8) | 9 (47.4) | 0.184 |
| IV | 1 (5.5) | 3 (15.8) | 0.340 |
Right atrial appendage (RAA) tissues weighing ~150
mg were removed at the beginning of the surgical interventions
under extracorporeal circulation. A portion of the tissues was
fixed in 4% paraformaldehyde for 24 h at 4°C and then embedded in
paraffin. The remainder of the tissues was snap-frozen in liquid
nitrogen for protein and RNA isolation.
In situ hybridization (ISH)
analysis
The expression of miR-208a-3p in RAA was
characterized by ISH with a double (3′ and 5′) digoxigenin
(DIG)-labelled locked nucleic acid modified probe
(5′-ACAAGCTTTTTGCTCGTCTTAT-3′; Exiqon, Vedbaek, Denmark).
Paraffin-embedded tissues sections were hybridized in 100 ml
hybridization buffer (Exiqon) with 50 pM denatured DIG-labelled
probe overnight at 42°C. The sections were then washed with 1X
saline-sodium phosphate EDTA buffer [8.765 g NaCl, 1.38 g
NaH2PO4 and 0.37 g
EDTA-Na2·2H2O dissolved in 500 ml diethyl
pyrocarbonate (DEPC)·H2O, pH 7.4] and incubated with an
anti-DIG-alkaline phosphatase antibody (cat. no. 11093274910;
Roche, Mannheim, Germany) diluted 1:2,000 in 1X blocking buffer (2%
sheep serum; cat no. 013-000-121; Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA) and 0.1% Triton X-100
dissolved with 100 mM Tris-HCl and 150 mM NaCl, pH 7.5 at room
temperature for 2 h. The sections were then stained with 45 µg
nitroblue tetrazolium in 10 ml of a solution of 50 ml Tris (pH
7.5), 10 ml 5 M NaCl and 25 ml 1 M MgCl2 in 500 ml
DEPC·H2O (pH 9.0) for 12 h and imaged under a light
microscope.
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from tissues and cells with
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The miRs were extracted from tissues with RNAiso
for Small RNA reagent (Takara Bio Inc., Otsu, Japan).
Total RNAs were reverse-transcribed into
complementary (c)DNA using a PrimerScript™ RT reagent kit with a
gDNA eraser (cat. no. RR047A, Takara Bio Inc.) according to the
manufacturer's protocols. The PCR mixture contained 2 µl cDNA as a
template, 10 µl SYBR® Premix Ex Taq (Takara Bio Inc.),
0.4 µl ROX Reference Dye (50X), 0.8 µl each of forward and reverse
primer (10 µM; Table II) and 6 µl
RNase-free deionized (d)H2O filled up to a final volume
of 20 µl. The PCR procedure was as follows: one cycle at 95°C for
30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 31
sec. Melting curve analysis was performed at 65–95°C. GAPDH was
used as the internal standard for normalizing gene expression.
| Table II.Primers of target genes. |
Table II.
Primers of target genes.
| Target gene | Primer | Base sequence |
|---|
| Cx40 | Sense |
5′-TCCTCGGAGTAGTGGTGAGATG-3′ |
|
| Antisense |
5′-AAAGCTGAGGCTGCTGGTAAAG-3′ |
| GAPDH | Sense |
5′-GCACCGTCAAGGCTGAGAAC-3′ |
|
| Antisense |
5′-TGGTGAAGACGCCAGTGGA-3′ |
| miR-208a-3p | Sense |
5′-ATAAGACGAGCAAAAAGCTTGT-3′ |
| U6 | Sense |
5′-GGAACGATACAGAGAAGATTAGC-3′ |
|
| Antisense |
5′-TGGAACGCTTCACGAATTTGCG-3′ |
The miRs were polyadenylated and subsequently
converted into cDNAs using a Mir-X™ miRNA First-Strand
Synthesis kit (Clontech Laboratories, Inc., Mountain View, CA, USA)
according to the manufacturer's protocols. The PCR mixture
contained 2 µl cDNA as the template, 12.5 µl SYBR Advantage Premix
(2X; Clontech Laboratories, Inc.), 0.5 µl ROX Reference Dye (50X),
0.5 µl of each of the mRQ 3′ Primer (Clontech Laboratories, Inc.)
and miR-specific 5′ primer (10 µM; Table II) and 9 µl RNase-free
dH2O to a final volume of 25 µl. The PCR procedure was
identical to that mentioned above. U6 small nuclear RNA (Clontech
Laboratories, Inc.) was used as the internal standard for
normalizing gene expression. All the data obtained were calculated
by the 2−ΔΔCq method (14).
Prediction of miR-208a-3p target
mRNAs
The target mRNAs of miR-208a-3p were predicted using
the following three databases: TargetScan (http://www.targetscan.org/), miRanda (http://www.microrna.org/microrna/home.do) and
RNAhybird (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid)
(15–17). Those consistently identified by the
three databases were regarded as potential target mRNAs. It was
demonstrated that GJA5 may be a potential target mRNA of
miR-208a-3p, as the latter exhibited binding to the 3′-untranslated
region (UTR) of Cx40 mRNA (Fig.
1).
Immunohistochemistry (IHC)
Paraffin-embedded tissues sections 4 µm thick were
routinely dewaxed in xylene and rehydrated in an ethanol gradient
series (100, 90, 80, 75 and 50%), followed by triple washing with
PBS. Antigen retrieval was performed at high pressure in sodium
citrate buffer (pH 6.0) at 125°C for 10 min. The sections were
blocked for 1 h at room temperature with endogenous peroxidase
blockers (cat no. PV-9000, Golden Bridge International, Inc.,
Bothell, WA, USA) and stained with anti-Cx40 antibody (cat. no.
ab38580 Abcam, Cambridge, UK) diluted at 1:200 overnight at 4°C.
Sections were then stained with polyperoxidase-conjugated
anti-mouse immunoglobulin (Ig) G (diluted according to the
manufacturer's protocol; cat. no. PV-9000, Golden Bridge
International, Inc.) for 1 h at room temperature. The signals were
then visualized using a diaminobenzidine kit (cat. no. ZLI-9017,
Zhongshan Goldenbridge Bio, Beijing, China) and observed under a
microscope.
Western blot analysis
Total protein was extracted from tissues and cells
using radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology, Shanghai, China) with phenylmethylsulfonyl fluoride
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). The protein
concentration was determined with a bicinchoninic acid protein
assay kit (Beyotime Institute of Biotechnology, Inc.). The proteins
were boiled with 4X SDS loading buffer (Takara Bio Inc.) and
separated by 10% SDS-PAGE, followed by transfer onto a 0.22-µm
polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA) using the Mini Trans-Blot electrophoretic transfer cell system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Membranes were
blocked for 0.5 h at room temperature with 5% non-fat milk in a
buffer of 20 mM Tris-HCl, 0.5 M NaCl and 0.1% Tween 20, and
incubated with anti-Cx40 antibody (Abcam) diluted at 1:1,000
overnight at 4°C. Subsequently, the membranes were incubated with
IRDye 800CW goat anti-mouse IgG (cat. no. 926-68071, LI-COR
Biotechnology, Inc., Lincoln, NE, USA) diluted at 1:10,000 for 1 h
at room temperature. The signals were detected and quantified with
the Odyssey system (LI-COR Biotechnology, Inc.). Protein band
intensities were expressed relative to β-tubulin, which was
incubated with β-tubulin antibody overnight at 4°C (1:10,000; cat
no. AF0524, Affinity Biologicals, Inc. Ancaster, ON, Canada).
Transfection with miR-208a-3p
inhibitor or mimics
To determine the effect of miR-208a-3p on Cx40
expression in cardiomyocytes, the AC16 cell line (18) (American Type Culture Collection,
Manassas, VA, USA) was treated with miR-208a-3p inhibitor or
mimics, respectively. The sequences were as follows: miR-208a-3p
inhibitor, 5′-ACAAGCUUUUUGCUCGUCUUAU-3′; miR inhibitor negative
control (NC), 5′-CAGUACUUUUGUGUAGUACAA-3′; miR-208a-3p mimics
sense, 5′-AUAAGACGAGCAAAAAGCUUGU-3′ and antisense,
5′-AAGCUUUUUGCUCGUCUUAUUU-3′; miR mimics NC sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. The AC16 cells were cultured in
24-well plates for 24 h and subsequently transfected with 80 nM
miR-208a-3p inhibitor, miR inhibitor NC, miR-208a-3p mimics or miR
mimics NC (Genepharma, Shanghai, China) using X-tremeGENE small
interfering (si)RNA Transfection reagent (Roche Diagnostics, Basel,
Switzerland) diluted in serum-free Opti-MEM®I medium
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocols. AC16 cells without siRNA and miR mimics and inhibitors
served as a blank control, it should be noted that the experiments
using miR inhibitors and mimics were not performed simultaneously,
which accounts for the different results for their respective blank
controls. The cells were collected for determination of Cx40
expression at 48 h after transfection.
Luciferase assay
To assess whether GJA5 was directly targeted by
miR-208a-3p, a luciferase assay was performed. The 293T cell line
(Cell Bank of the Chinese Academy of Sciences, Shanghai, China) was
cultured in 24-well plates for 24 h prior to transfection.
Subsequently, the cells were co-transfected with 0.8 µg wild-type
or mutant-type GJA5-3′UTR of GJA5 mRNA luciferase reporter plasmid
(Genepharma) and 40 nM miR-208a-3p mimics or miR mimics NC
(Genepharma) using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) diluted in Opti-MEM®I reduced serum
medium (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocols.
Firefly and Renilla luciferase activities were
measured at 48 h after transfection using the Dual Luciferase Assay
System (Promega Corp., Madison, WI, USA), according to the
manufacturer's protocols. Firefly luciferase activity was
normalized to that of Renilla.
Statistical analysis
All quantitative data are expressed as the mean ±
standard deviation. Statistical significance between 2 groups was
assessed by Student's-test and among multiple groups using one-way
analysis of variance followed by the least-significant differences
post hoc test. P<0.05 was considered to indicate a statistically
significant difference. All statistical analyses were performed
using SPSS 16.0 (SPSS Inc., Chicago, IL, USA).
Results
miR-208a-3p is upregulated in RAA of
patients with AF
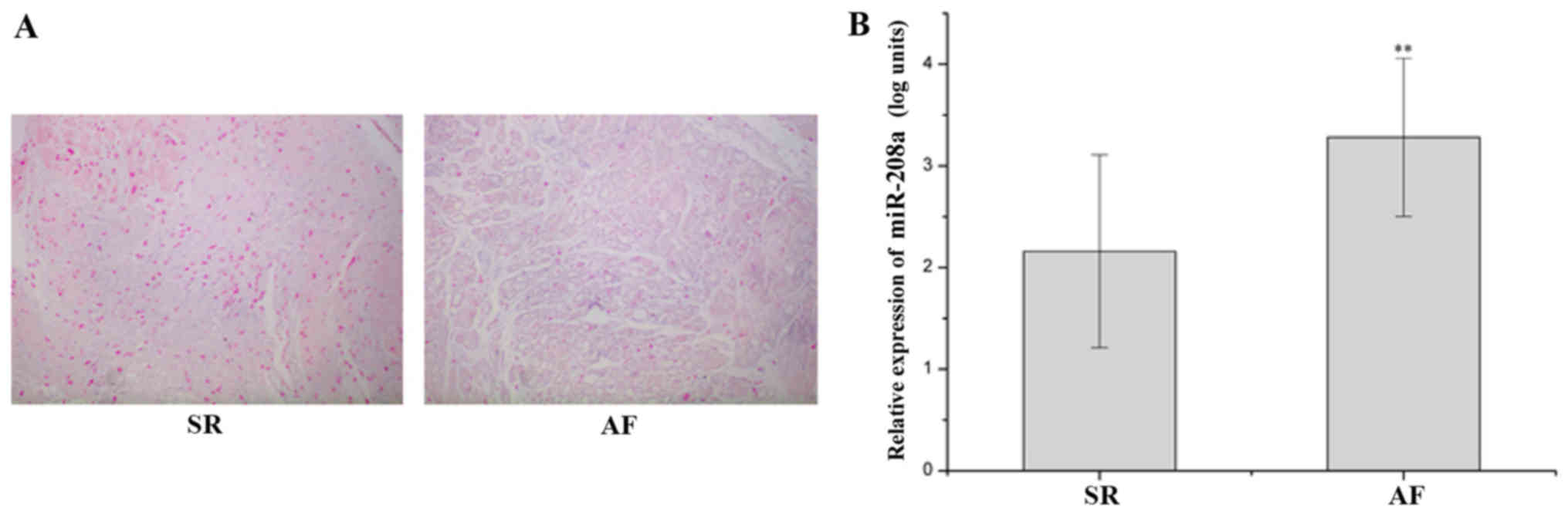
A low expression of miR-208a-3p, which was
distributed in the cardiomyocytes and cardiac interstitial spaces,
was observed in the SR group, and miR-208a-3p expression was
elevated in the AF group as indicated by ISH (Fig. 2A). The miR-208a-3p levels in the AF
group were significantly upregulated compared with those in the SR
group (P<0.01; Fig. 2B).
Lateralization and decreased
expression of Cx40 in RAA of patients with AF
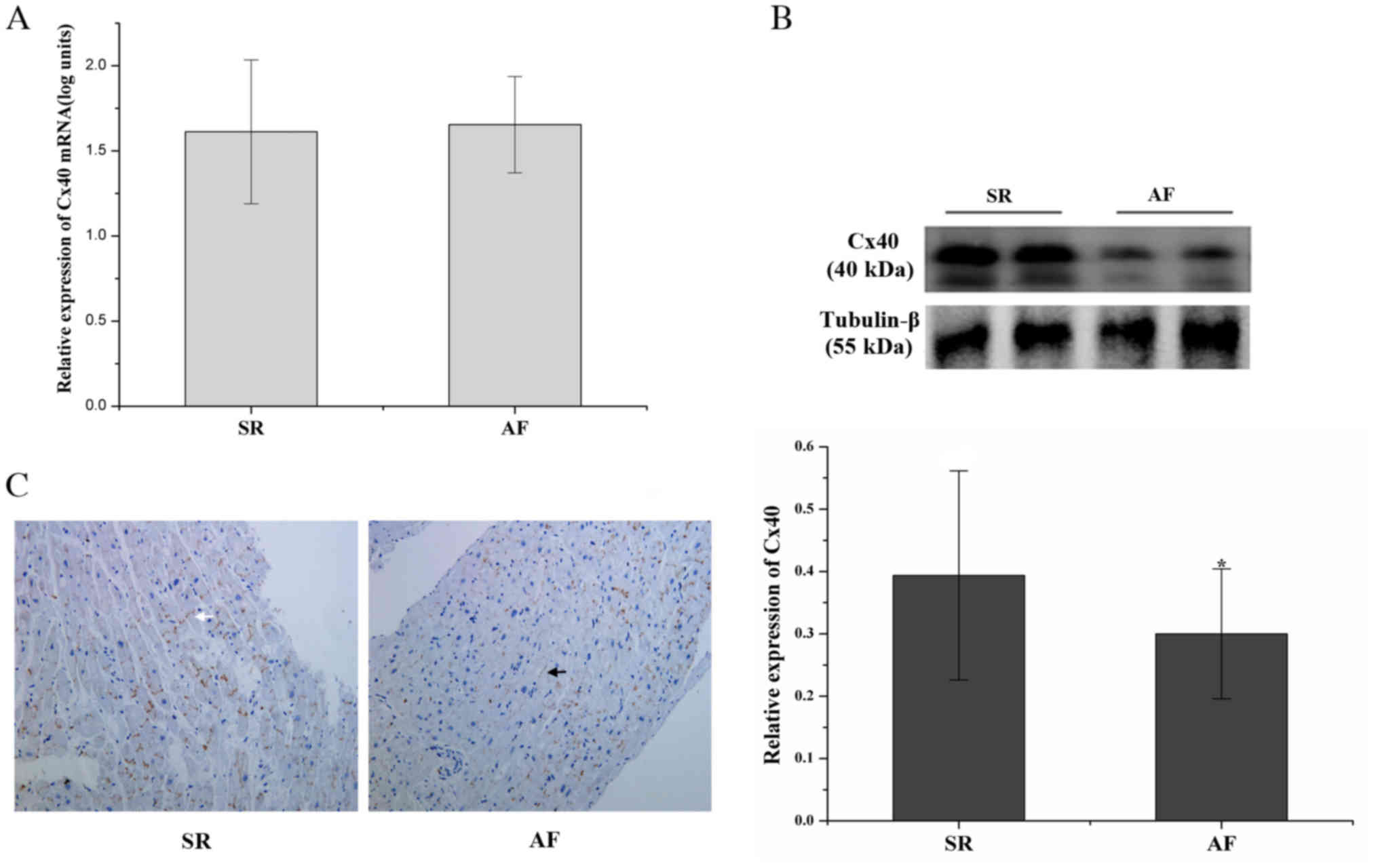
Although no significant difference in Cx40 mRNA
levels was identified between the AF and SR groups (P>0.05;
Fig. 3A), the Cx40 protein levels in
the AF group were decreased by 23.81% compared with those in the SR
group (P<0.05; Fig. 3B).
The expression of Cx40 in RAA was characterized by
IHC. Cx40 was predominantly present at end-to-end connections
between cardiomyocytes in the SR group. The presence of Cx40 at
side-to-side connections between cardiomyocytes was markedly
increased in the AF group, and reduced expression of Cx40 was
observed as well (Fig. 3C).
miR-208a-3p negatively regulates Cx40
expression in AC16 cells, but does not directly target GJA5
As human AC16 cell line exhibits numerous
biochemical and morphological characteristics of cardiac muscle
cells (18), it was used in the
present study to assess whether Cx40 was regulated by miR-208a-3p
in human cardiomyocytes. The expression of Cx40 was determined in
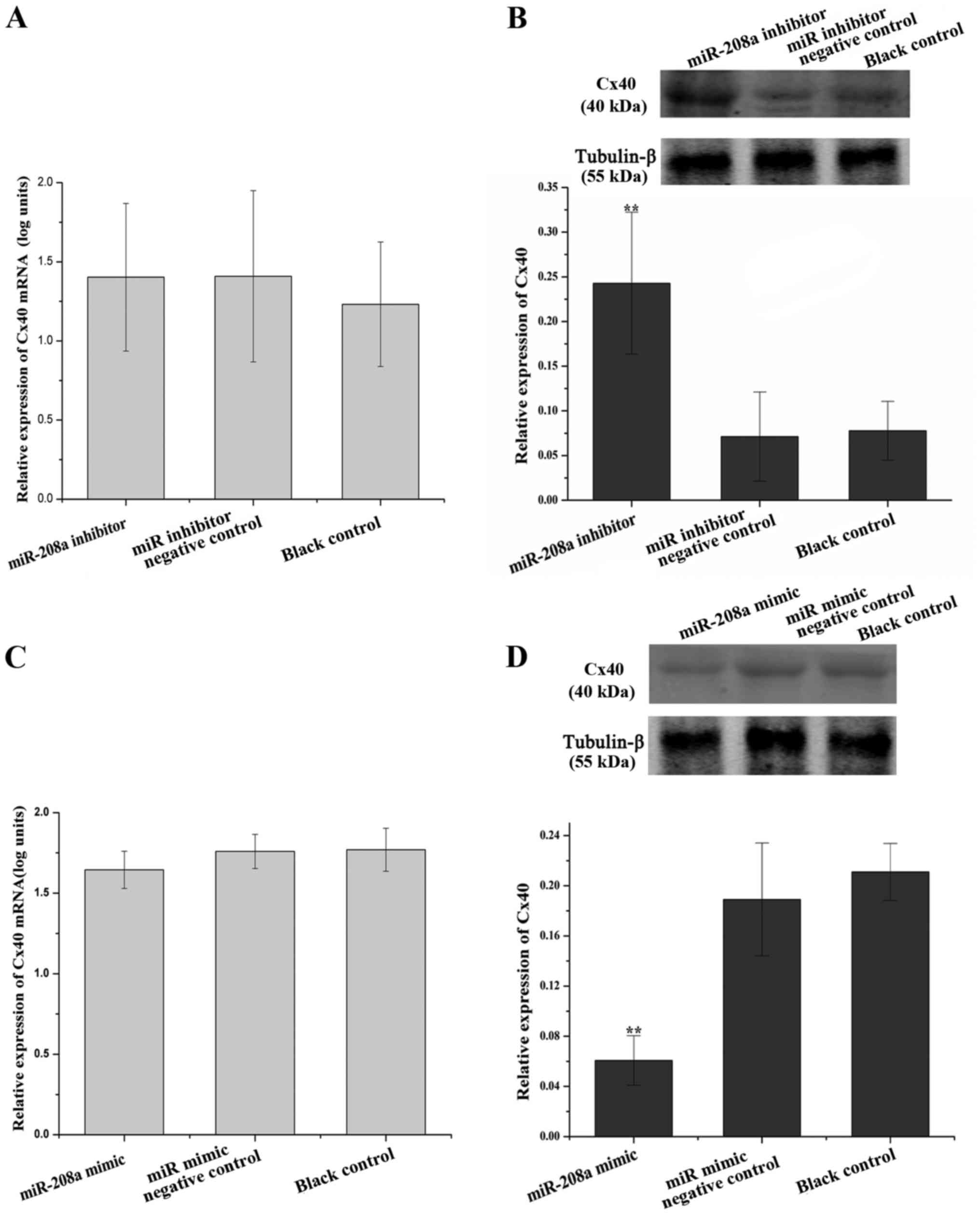
AC16 cells treated with miR-208a-3p inhibitor or mimics. The Cx40
mRNA levels were not significantly different among the miR-208a-3p
inhibitor group, the miR inhibitor NC group and the blank control
group (P>0.05; Fig. 4A). However,
the Cx40 protein levels in the miR-208a-3p inhibitor group were
upregulated 3.41-fold compared with those in the miR inhibitor NC
group (P<0.01; Fig. 4B), but
there was no significant difference between the miR inhibitor NC
and the blank control group (P>0.05; Fig. 4B). The Cx40 mRNA levels were not
significantly different among the miR-208a-3p mimics group, the miR
mimics NC group and the blank control group (P>0.05; Fig. 4C). However, the Cx40 protein levels
in the miR-208a-3p mimics group was downregulated 3.11-fold
compared with that in the miR mimics NC group (P<0.01; Fig. 4D), while there was no significant
difference between the miR mimics NC and the blank control group
(P>0.05; Fig. 4D). These results
demonstrated that miR-208a-3p is an upstream negative regulator of
Cx40.
To confirm whether GJA5 was directly targeted by
miR-208a-3p, a luciferase assay was performed. However,
overexpression of miR-208a-3p did not significantly inhibit the
luciferase activity of the reporter plasmid carrying the wild-type
3′-UTR sequence of GJA5 (P>0.05; Fig.
5).
The aforementioned results indicate that although
miR-208a-3p did not target GJA5 directly, it was responsible for
Cx40 remolding. The signaling pathways via which miR-208a-3p
regulates Cx40 expression and remolding require further
investigation.
Discussion
Numerous lines of evidence have indicated that
variants in GJA5 and its promoter sequence are associated with a
predisposition to AF (19–21). Reduced expression and lateralization
of Cx40 were identified in atriums of patients with AF and animal
models of AF, followed by decreased speed and increased
heterogeneity of conduction (22–24). In
addition, decelerated atrial conduction and increased vulnerability
to atrial arrhythmias were observed in a Cx40-deficient mouse model
(25). The present study identified
a downregulation of Cx40 protein levels and lateralization of Cx40
in RAA tissues of patients with AF compared with that in patients
with SR. These results indicated that Cx40 remolding is an
important contributor to AF.
miR-208 is crucial for the development of
cardiomyocytes and the cardiac conduction system (26). Previous studies revealed that miR-208
was involved in various cardiac pathological conditions, including
cardiac fibrosis, cardiac hypertrophy, dilated cardiomyopathy and
heart failure (27–29). Recently, miR-208 was identified to be
closely associated with AF in animal models and patients. First- or
second-degree atrioventricular block (AVB) was identified in
miR-208a-3p transgenic mice and the majority of miR-208 knockout
mice displayed first-degree AVB and AF (30). Nishi et al (11) reported that miR-208b in the right
atrium of patients with AF was elevated compared with that in
patients with SR. Radtke et al (12) identified an increase in miR-208a-3p
expression in the left atriums from paroxysmal in persistent AF.
The present study detected an elevated expression of miR-208a-3p in
RAA tissues of patients with AF. It is suggested that the
upregulation of miR-208a-3p may promote AF by decelerating atrial
conduction (30).
In the present study, a negative association between
miR-208a-3p and Cx40 protein levels was identified. Of note, the
3′-UTR of Cx40 mRNA contained a predicted miR-208a-3p target site.
In addition, the Cx40 mRNA levels were not significantly different
between the SR and the AF groups. Therefore, it was speculated that
miR-208a-3p repressed GJA5 expression at the post-transcriptional
level by binding to the 3′-UTR of Cx40 mRNA directly. To
experimentally verify this, AC16 cells were treated with
miR-208a-3p inhibitor or mimics. The results indicated that the
Cx40 transcript levels in AC16 cells treated with miR-208a-3p
inhibitor or mimics were similar. However, the Cx40 protein levels
were significantly upregulated by miR-208a-3p inhibitor and
conversely, treatment with miR-208a-3p mimics significantly
downregulated the Cx40 protein expression. These results further
suggested that miR-208a-3p may be involved in AF by targeting Cx40
mRNA directly. Subsequently, a luciferase assay was performed to
assess whether GJA5 was directly suppressed by miR-208a-3p.
However, co-transfection of miR-208a-3p with the GJA5-3′UTR
luciferase reporter vector did not suppress luciferase activity.
This result indicated that the negative regulatory effect of
miR-208a-3p on Cx40 was not achieved by targeting GJA5 directly.
Taken together, these findings demonstrated that miR-208a-3p is
responsible for AF by indirectly promoting Cx40 remolding, and it
is an important upstream negative regulatory factor of Cx40 in
human chronic AF.
To the best of our knowledge, the present study was
the first to report that miR-208a-3p contributes to Cx40 remolding
in human chronic AF. However, the present study had certain
limitations. First, the expression changes of miR-208a-3p and Cx40
were only detected in valvar heart disease patients with
long-standing, persistent AF. Patients with other types of
cardiovascular disease and those with AF of a different duration
require to be examined in the future. Furthermore, the present
findings only proved that miR-208a-3p is an upstream negative
regulatory factor of Cx40 in human chronic AF. The specific
molecular mechanisms underlying miR-208a-3p-mediated Cx40 remolding
require further elucidation. Callis et al (30) reported decreased Cx40 expression in
miR-208a-3p knockout mice. They suggested that downregulated Cx40
expression partially resulted from reduced heat shock protein (HSP)
70-Hsp90 organizing protein (Hop) expression. However, as Hop was
not directly regulated by miR-208a-3p, they speculated that the
reduced Hop expression might stem from increased expression of
GATA4, which is targeted by miR-208a-3p directly, but the potential
mechanisms of Hop downregulation induced by GATA4 remained elusive.
GATA4 is a cardiac transcription factor, which transactivates the
Cx40 promoter (31). Therefore, it
may be speculated that Cx40 remolding mediated by miR-208a-3p may
be partially associated with downregulation of GATA4 in human AF.
However, the mRNA levels of Cx40 were not affected in patients with
AF and in AC16 cells treated with miR-208a-3p inhibitor or mimics.
This indicated that the mechanisms underlying miR-208a-3p-mediated
Cx40 remolding are involved in a complex regulatory network at the
transcriptional and post-transcriptional level. Finally, the
present study clarified the negative regulatory effect of
miR-208a-3p on Cx40 in vitro. This effect requires to be
further verified in vivo.
miR-208a-3p is an important upstream negative
regulatory factor of Cx40 and the upregulation of miR-208a-3p was
indicated to be responsible for Cx40 remolding in human chronic AF.
This may represent a potential therapeutic target for AF. However,
the specific mechanisms underlying miR-208a-3p-mediated Cx40
remolding require further elucidation.
Acknowledgements
The authors are grateful to Professor Shasha Liu
(English Language Department, Guangxi Medical University, Nanning,
China) for helping to revise and edit the manuscript. The present
study was supported by the National Natural Science Foundation of
China (grant no. 81460057).
References
|
1
|
Rahman F, Kwan GF and Benjamin EJ: Global
epidemiology of atrial fibrillation. Nat Rev Cardiol. 11:639–654.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nattel S and Dobrev D:
Electrophysiological and molecular mechanisms of paroxysmal atrial
fibrillation. Nat Rev Cardiol. 13:575–590. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wakili R, Voigt N, Kääb S, Dobrev D and
Nattel S: Recent advances in the molecular pathophysiology of
atrial fibrillation. J Clin Invest. 121:2955–2968. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bikou O, Thomas D, Trappe K, Lugenbiel P,
Kelemen K, Koch M, Soucek R, Voss F, Becker R, Katus HA and Bauer
A: Connexin 43 gene therapy prevents persistent atrial fibrillation
in a porcine model. Cardiovasc Res. 92:218–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Segretain D and Falk MM: Regulation of
connexin biosynthesis, assembly, gap junction formation, and
removal. Biochim Biophys Acta. 1662:3–21. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Davis LM, Kanter HL, Beyer EC and Saffitz
JE: Distinct gap junction protein phenotypes in cardiac tissues
with disparate conduction properties. J Am Coll Cardiol.
24:1124–1132. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Igarashi T, Finet JE, Takeuchi A, Fujino
Y, Strom M, Greener ID, Rosenbaum DS and Donahue JK: Connexin gene
transfer preserves conduction velocity and prevents atrial
fibrillation. Circulation. 125:216–225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Rooij E and Olson EN: MicroRNA
therapeutics for cardiovascular disease: Opportunities and
obstacles. Nat Rev Drug Discov. 11:860–872. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo X, Yang B and Nattel S: MicroRNAs and
atrial fibrillation: Mechanisms and translational potential. Nat
Rev Cardiol. 12:80–90. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nattel S and Harada M: Atrial remodeling
and atrial fibrillation: Recent advances and translational
perspectives. J Am Coll Cardiol. 63:2335–2345. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nishi H, Sakaguchi T, Miyagawa S,
Yoshikawa Y, Fukushima S, Saito S, Ueno T, Kuratani T and Sawa Y:
Impact of microRNA expression in human atrial tissue in patients
with atrial fibrillation undergoing cardiac surgery. PLoS One.
8:e733972013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Radtke A, Hanke T, Yan J, Godau B, Cordes
J, Nigam V, Sievers HH and Mohamed SA: MicroRNA 208 in atrial
fibrillation. J Clin Exp Cardiol. 5:3252014.
|
|
13
|
January CT, Wann LS, Alpert JS, Calkins H,
Cigarroa JE, Cleveland JC Jr, Conti JB, Ellinor PT, Ezekowitz MD,
Field ME, et al: 2014 AHA/ACC/HRS guideline for the management of
patients with atrial fibrillation: A report of the American College
of Cardiology/American Heart Association Task Force on Practice
Guidelines and the Heart Rhythm Society. J Am Coll Cardiol.
64:e1–e76. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:2015. View Article : Google Scholar
|
|
16
|
Betel D, Koppal A, Agius P, Sander C and
Leslie C: Comprehensive modeling of microRNA targets predicts
functional non-conserved and non-canonical sites. Genome Biol.
11:R902010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rehmsmeier M, Steffen P, Hochsmann M and
Giegerich R: Fast and effective prediction of microRNA/target
duplexes. RNA. 10:1507–1517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Davidson MM, Nesti C, Palenzuela L, Walker
WF, Hernandez E, Protas L, Hirano M and Isaac ND: Novel cell lines
derived from adult human ventricular cardiomyocytes. J Mol Cell
Cardiol. 39:133–147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Firouzi M, Ramanna H, Kok B, Jongsma HJ,
Koeleman BP, Doevendans PA, Groenewegen WA and Hauer RN:
Association of human connexin40 gene polymorphisms with atrial
vulnerability as a risk factor for idiopathic atrial fibrillation.
Circ Res. 95:e29–e33. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gollob MH, Jones DL, Krahn AD, Danis L,
Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, et al:
Somatic mutations in the connexin 40 gene (GJA5) in atrial
fibrillation. N Engl J Med. 354:2677–2688. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Juang JM, Chern YR, Tsai CT, Chiang FT,
Lin JL, Hwang JJ, Hsu KL, Tseng CD, Tseng YZ and Lai LP: The
association of human connexin 40 genetic polymorphisms with atrial
fibrillation. Int J Cardiol. 116:107–112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kostin S, Klein G, Szalay Z, Hein S, Bauer
EP and Schaper J: Structural correlate of atrial fibrillation in
human patients. Cardiovasc Res. 54:361–379. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rucker-Martin C, Milliez P, Tan S, Decrouy
X, Recouvreur M, Vranckx R, Delcayre C, Renaud JF, Dunia I,
Segretain D and Hatem SN: Chronic hemodynamic overload of the atria
is an important factor for gap junction remodeling in human and rat
hearts. Cardiovasc Res. 72:69–79. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiao P, Gao C, Fan J, Du H, Long Y and Yin
Y: Blockade of angiotensin II improves hyperthyroid induced
abnormal atrial electrophysiological properties. Regul Pept.
169:31–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hagendorff A, Schumacher B, Kirchhoff S,
Lüderitz B and Willecke K: Conduction disturbances and increased
atrial vulnerability in Connexin40-deficient mice analyzed by
transesophageal stimulation. Circulation. 99:1508–1515. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oliveira-Carvalho V, Carvalho VO and
Bocchi EA: The emerging role of miR-208a in the heart. DNA Cell
Biol. 32:8–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van Rooij E, Sutherland LB, Qi X,
Richardson JA, Hill J and Olson EN: Control of stress-dependent
cardiac growth and gene expression by a microRNA. Science.
316:575–579. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Satoh M, Minami Y, Takahashi Y, Tabuchi T
and Nakamura M: Expression of microRNA-208 is associated with
adverse clinical outcomes in human dilated cardiomyopathy. J Card
Fail. 16:404–410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paulin R, Sutendra G, Gurtu V, Dromparis
P, Haromy A, Provencher S, Bonnet S and Michelakis ED: A
miR-208-Mef2 axis drives the decompensation of right ventricular
function in pulmonary hypertension. Circ Res. 116:56–69. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Callis TE, Pandya K, Seok HY, Tang RH,
Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, et al:
MicroRNA-208a is a regulator of cardiac hypertrophy and conduction
in mice. J Clin Invest. 119:2772–2786. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Linhares VL, Almeida NA, Menezes DC,
Elliott DA, Lai D, Beyer EC, de Carvalho Campos AC and Costa MW:
Transcriptional regulation of the murine Connexin40 promoter by
cardiac factors Nkx2-5, GATA4 and Tbx5. Cardiovasc Res. 64:402–411.
2004. View Article : Google Scholar : PubMed/NCBI
|