Introduction
Colorectal cancer (CRC) is the second leading cause
of cancer-related mortality in Asia, and is classified as a
terminal illness due to its relatively high rates of
chemoresistance and hepatic metastasis (1). To overcome these problems, novel
strategies in addition to primary treatments for colorectal cancer
have been investigated (2). At
present, primary treatment for CRC involves complete surgical
removal of the primary tumor and regional lymph nodes (3). The prognosis of patients with CRC is
dependent on tumor stage and grade; however, as resection is
typically performed in late stage CRC for 30–40% of cases, the
prognosis is generally poor (4).
Hyperthermia exposure above temperatures that are
physiologically optimal affects numerous biological processes in
cells and tissues, including proliferation, migration, invasion and
apoptosis, by modifying the physical properties of cellular
components to induce negative cellular responses (5). Hyperthermia is typically studied in
combination with chemotherapy drugs. Fałkowska-Podstawka and
Wernicki (6) have reported that
hyperthermia exposure denatures proteins and induces DNA and RNA
damage, which interrupts vital cellular processes and promotes
apoptosis. Kim et al (7)
identified a potential mechanism of apoptotic induction following
hyperthermia exposure that involved activation of tumor necrosis
factor-related apoptosis-inducing ligand. Additionally, Kalamida
et al (8) reported that
hyperthermia not only potently induced apoptosis by activating
caspase-9, but also exerted a universal suppressive effect on
cancer cell proliferation and reversed chemoresistance caused by
chemoagent treatment. The potential mechanisms regarding inhibited
proliferation are considered to involve the suppression of tumor
cell replication and metabolism. For instance, in the renal cell
carcinoma (RCC) cell line 786-O, hyperthermia exposure suppressed
Ku80 expression and lead to G2/M phase cell cycle arrest, which may
represent a potential mechanism of hyperthermia-mediated
suppression of proliferation (9).
The cellular response to hyperthermia has been
widely studied, though the role of microRNA (miRNA/miR) in
hyperthermia remains largely unknown. A number of miRNAs have been
demonstrated to be thermally responsive (10,11). Due
to the numerous functions of miRNAs, studies into the miRNA profile
associated with hyperthermia are necessary. Notably, hyperthermic
miRNAs may be mediators of some of the biological and/or
therapeutic properties of hyperthermia (12).
The gene for p53 (TP53) is mutated in 50% of human
cancers, including CRC (13). p53 is
among the most well-established cancer inhibitors, and regulates
cell cycle arrest, DNA repair and apoptosis (14). The effect of hyperthermia on p53
expression indicates an involvement of p53 in hyperthermic
inhibition of tumor growth (15).
Furthermore, hyperthermia exposure activated p53 and caused the
activation of cell cycle checkpoints (16). They also reported that, in human
liver carcinoma HepG2 cells expressing wild type p53, hyperthermia
caused DNA damage response-induced G2/M arrest through activated
p53 and ATM/ATR, which was followed by apoptosis, while this was
not observed in Huh7 cells expressing mutant p53 (16). These results indicate a critical role
of p53 in the cellular effects of hyperthermia.
Multiple miRNAs, including miR-215, miR-504, and
miR-34a, b and c, may be transcriptionally activated by p53
(17). Furthermore, the miR-34a has
also been reported to serve a critical regulatory role in the
posttranscriptional functions of p53 (18). Thus, it may be hypothesized that
feedback regulation exists between miR-34 and p53.
In the present study, it was determined whether
hyperthermia could induce apoptosis and cell cycle arrest in the
CRC cell line HCT116, and the involvement of p53 was investigated.
The potential regulatory effects of microRNA-34a in the
p53-dependent hyperthermic response were also assessed.
Materials and methods
Cell culture and treatment
Human colorectal cancer HCT116 and TP53-/- HCT116
cells (HCT116TP53-/−) cells were purchased from the
American Type Culture Collection (Manassas, VA, USA). The cells
were maintained in Dulbecco's modified Eagle's medium (Thermo
Fisher Scientific, Inc., Waltham, MA, USA), supplemented with 10%
heat-inactivated fetal bovine serum (FBS, Gibco; Thermo Fisher
Scientific, Inc.) at 37°C and 5% CO2. The medium was
replenished every 3 days and when the cells reached 80–90%
confluence, they were suspended with 0.25% trypsin (Gibco; Thermo
Scientific, Inc.) and used in experiments.
In a hyperthermia exposure group (HYP),
5×104/well HCT116 cells seeded in 12-well plates were
incubated in DMEM medium supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) in an incubator (Forma Series II 3110;
Thermo Fisher Scientific, Inc.) preheated to 42°C for 0, 2 and 4 h.
Additionally, a HYP group of HCT116TP53-/− cells was
subjected to hyperthermia for 2 h. Control groups (CTL) of each
cell line were incubated at 37°C for the same time periods.
Following hyperthermia exposure, the cells were incubated at 37°C
for the indicated time periods (2 and 4 h) prior to analysis.
Transfection with microRNA mimics
Oligofectamine reagent (Thermo Fisher Scientific,
Inc.), miR-34a (cat. no. C-300551-07; GE Healthcare Dharmacon,
Inc., Lafayette, CO, USA), b (Assay ID, PM10743; cat. no. AM17100)
and c (Assay ID, AM12342; cat. no. AM17000), scrambled mimics (cat.
no. 4464058), and antago-scrambled (cat. no. 4464076) and
antago-miR-34a miRNAs (Assay ID, MH11030; cat. no. 4464084) (all
Thermo Fisher Scientific, Inc.) were employed following the
manufacturer's protocol. To detect the effect of miR-34a, b or c on
the transcriptional activity of p53, miR-34a, b or c, or negative
control scrambled mimics were transfected into HCT116 cells. To
detect the effects of miR-34a on apoptosis and cell cycle
distribution, antago-miR-34a or antago-scrambled miRNAs were
transfected into HCT116 cells. Briefly, 5×105 cells/well
were plated in a 6-well plate (Corning Incorporate, Corning, NY,
USA) for 16 h. A total of 200 nM microRNA mimics was transfected
transiently into cells. Flow cytometry and luciferase reporter
assay experiments were performed 24 h after transfection. Western
blotting and reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) analysis were performed 48 h after
transfection.
Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) double staining and flow cytometry
analysis
The HCT116 cells were subjected to hyperthermia or
normothermia as above for 0–4 h. Subsequently, an annexin V-FITC/PI
double staining assay was performed according to the manufacturer's
protocol of an Annexin V-FITC/PI apoptosis detection kit (Thermo
Fisher Scientific, Inc.). In brief, ~5×105 cells were
harvested, washed with chilled PBS and resuspended in binding
buffer containing 5 µl Annexin V-FITC for 10 min in the dark at
room temperature, after which the binding buffer was removed by
centrifugation at 1,000 × g at 4°C for 10 min. The cells were then
resuspended in reaction buffer containing 5 µl PI, and flow
cytometry analysis was immediately performed to detect apoptosis
using a three laser Navios flow cytometer (Beckman Coulter, Inc.,
Brea, CA, USA) according the manufacturer's protocol and FlowJo
software (version 7.6.3; FlowJo LLC, Ashland, OR, USA)
software.
Cell cycle analysis
The cells (1×106) were washed with
ice-cold PBS, collected and fixed in 70% ice-cold ethanol overnight
at 4°C and incubated with with 5 µg/ml PI Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) at 37°C for 10 min, subsequently cell
cycle analyses were performed using a three laser Navios flow
cytometer according the manufacturer's protocol and FlowJo software
(version 7.6.3). The proportion of cells in G0/G1, S, and G2/M
phases was measured.
Luciferase reporter assays
A pGL3 firefly luciferase reporter plasmid
containing the p53 consensus binding sequence (pGL3-P53BS,
5′-TACAGAACATGTCTAAGCATGCTGGGGACT-3′ 30 mer) (19). A control pGL3-basic vector and a
Renilla luciferase internal control plasmid, pRL-SV40, were
purchased from Promega Corporation (Madison, WI, USA). A total of
1×105 cells in each group were cultured in 24-well
plates in DMEM containing 10% FBS at 37°C overnight. The cells were
cotransfected with pRL-SV40 and pGL3-P53BS or pGL3-basic using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. The cells were harvested
24 and 48 h after transfection and cell lysates were analysed for
firefly and renilla luciferase activity using a
Dual-Luciferase® Reporter Assay System (Promega
Corporation), according to the manufacturer's protocol, and a
microplate reader (Synergy 2 Multi-Mode Microplate Reader; BioTek
Instruments, Inc., Winooski, VT, USA). Data were presented as a
fold induction in luciferase activity compared with the pGL3-basic
vector. Three independent transfection experiments were performed
in triplicate for each experimental construct.
Western blot analysis
Cells were subjected to hyperthermia or normothermia
as above. The cells were then harvested, washed twice with 1 ml
chilled PBS, and lysed with total protein lysis buffer (Guangzhou
RiboBio Co., Ltd., Guangzhou, China). Total protein (50 µg)
measured by bicinchoninic acid assay kit (Sigma-Aldrich; Merck
KGaA) was mixed with 10 µl 5X SDS loading buffer and incubated for
10 min at 100°C, and then 50 µg total protein was separated by 12%
SDS-PAGE and transferred to polyvinylidene fluoride (PVDF)
membranes. After transferring, 5% bovine serum albumin
(Sigma-Aldrich; Merck KGaA) in PBS was employed as a blocking
solution to block unspecific sites on the PVDF membranes at room
temperature for 30 min. Western blotting was performed with
anti-β-actin antibody at 1:5,000 dilution (cat. no. mAbcam 8226;
Abcam, Cambridge, UK), anti-pro caspase 3 antibody at 1:1,000
dilution (cat. no. ab32150; Abcam) and anti-active caspase 3
antibody at 1:1,000 dilution (cat. no. ab32042; Abcam) at room
temperature for 2 h. A horseradish peroxidase-conjugated
anti-rabbit immunoglobulin G antibody at 1:5,000 dilution (cat. no.
ab6721; Abcam) was used as the secondary antibody at room
temperature for 1 h. β-actin was used as the internal reference.
Protein signals were detected using enhanced chemiluminescence
reagents (Thermo Fisher Scientific, Inc.).
RT-qPCR analysis
Total RNA was extracted from the HCT116 and
HCT116TP53-/− cells exposed to hyperthermia or
normothermia for 2 h using TRIzol reagent (Thermo Fisher
Scientific, Inc.), according to the manual. Following RNA
isolation, the concentration of purified RNA was determined with a
UV spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA was
reverse transcribed from the extracted total RNA using a Reverse
Transcriptase kit (Guangzhou RiboBio Co., Ltd.). To analyse miRNA
levels, RT was performed with specific miRNA stem-loop primers for
miR-34a (assay ID, 478047_mir), miR-34b (assay ID, 478050_mir),
miR-34c (assay ID, 478052_mir), miR-215 (assay ID, 478516_mir) and
miR-504 (assay ID, 478956_mir) (all Thermo Fisher Scientific,
Inc.). The PCR was performed using Taqman® Universal PCR
Master mix kit (Thermo Fisher Scientific, Inc.) and the ABI7500
system (Applied Biosystems; Thermo Fisher Scientific, Inc.) under
the following conditions: 95°C for 10 min, then 60 cycles of 95°C
for 15 sec and 60°C for 1 min. The levels of miRNA were measured
using the primer-probes from the Taqman microRNA Assay system (cat.
no. A25576; Applied Biosystems; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. U6 small nuclear RNA
(Assay ID, 001973; cat. no. 4427975; Thermo Fisher Scientific,
Inc.), provided by the Taqman microRNA Assay system, was used for
normalization.
To analyze mRNA levels, a SYBR-Green Master Mix
(Thermo Fisher Scientific, Inc.) was used in the ABI7500 system
under the following conditions: 95°C for 10 min, then 60 cycles of
95°C for 15 sec and 60°C for 1 min The specific primers used were
as follows: For Forlow, forward, 5′-CATGTACGTTGCTATCCAGGC-3′ and
reverse, 5′-CTCCTTAATGTCACGCACGAT-3′; for p21, forward,
5′-TGTCCGTCAGAACCCATGC-3′ and reverse, 5′-AAAGTCGAAGTTCCATCGCTC-3′;
for B cell lymphoma 2-associated X protein (BAX), forward,
5′-CCCGAGAGGTCTTTTTCCGAG-3′ and reverse,
5′-CCAGCCCATGATGGTTCTGAT-3′; for mouse double minute 2 homolog
(MDM2), forward, 5′-ACGACAAAGAAAACGCCACA-3′ and reverse,
5′-CTCTCCCCTGCCTGATACAC-3′; for p53 upregulated modulator of
apoptosis (PUMA), forward, 5′-GCCAGATTTGTGAGACAAGAGG-3′ and
reverse, 5′-CAGGCACCTAATTGGGCTC-3′; and for growth arrest and
DNA-damage-inducible 45α (GADD45A), forward,
5′-GAGAGCAGAAGACCGAAAGGA-3′ and reverse,
5′-CACAACACCACGTTATCGGG-3′. β-actin mRNA expression was used to
normalize the mRNA levels. All data was calculated using the
comparative threshold cycle (2−ΔΔCq) method (20).
Statistical analysis
All data were analyzed for statistical significance
using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). Data were
expressed as the mean ± standard deviation from at least 3
independent experiments performed in duplicate. Statistical
comparisons of the results were performed using one-way analysis of
variance followed by a Tukey's post hoc test. P<0.05 was
considered to indicate statistical significance.
Results
Effects of hyperthermia on cell
cycling and apoptosis in HCT116 cells
To assess whether hyperthermia induced cell cycle
arrest and apoptosis in HCT116 cells, the cells were maintained at
37°C (CTL group) or 42°C (HYP group) for three time intervals (0, 2
and 4 h), respectively. The cells were then analyzed by flow
cytometry to determine the rate of apoptosis and percentages of
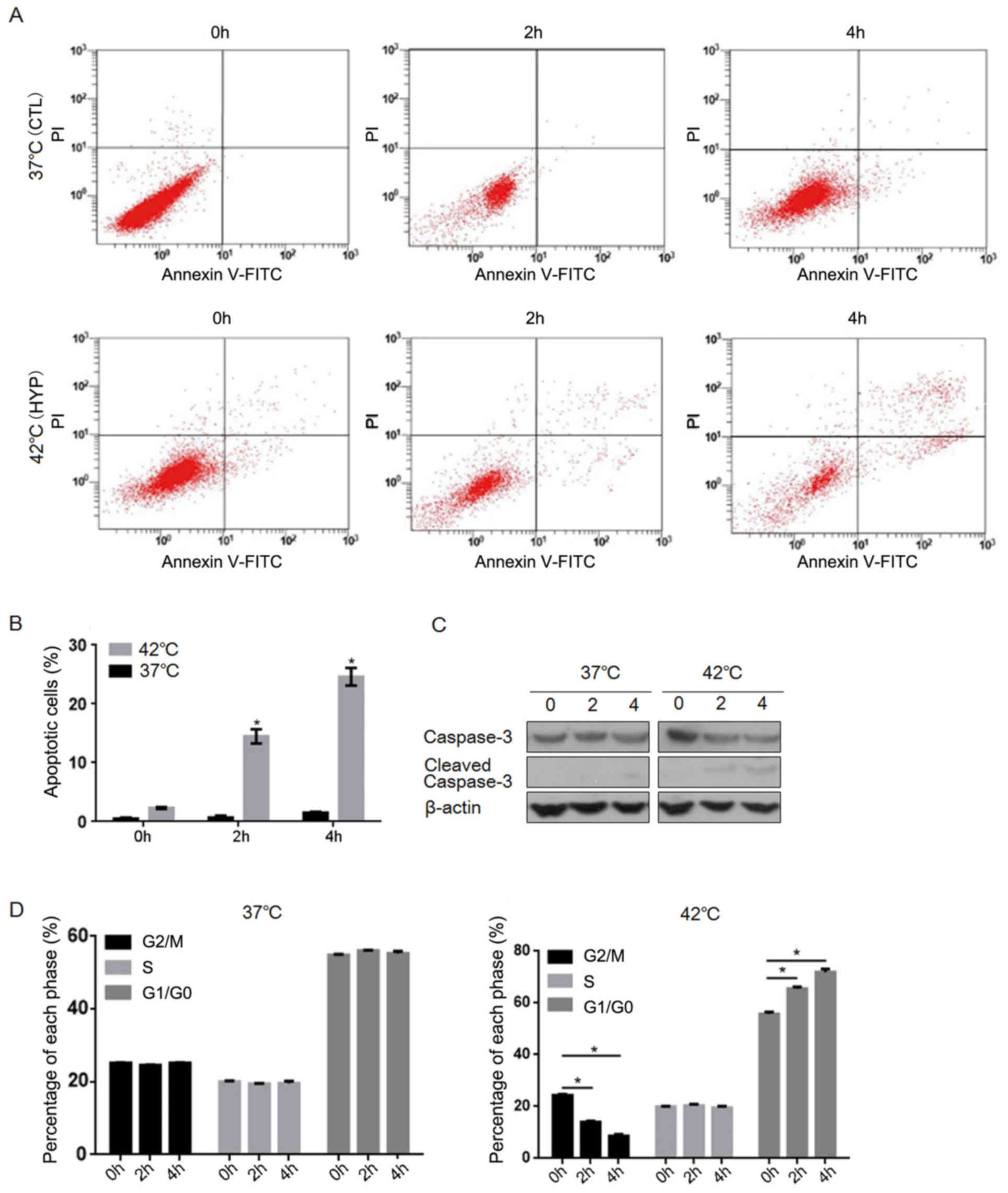
different cell phases. As depicted in Fig. 1A and B, no significant change in the
rate of cell apoptosis was observed in the CTL group following
incubation at 37°C for the different time intervals. By contrast,
the percentages of apoptotic cells in the HYP group significantly
increased following incubation at 42°C for 2 and 4 h (P<0.05 vs.
0 h HYP group; Fig. 1A and B). To
verify that this elevation in apoptosis was caused by hyperthermia
exposure, the levels of cleaved caspase-3 were assessed by western
blotting. As depicted in Fig. 1C, it
was observed that hyperthermia exposure increased the levels of
cleaved caspase-3 over time. Additionally, the results of cell
cycle analysis demonstrated that the percentages of G1/G0-phase
cells in the HYP group significantly increased, while those of
G2/M-phase cells significantly decreased, following hyperthermia
exposure for 2 and 4 h (P<0.05; Fig.
1D). These results demonstrated that hyperthermia significantly
induced apoptosis and cell cycle arrest in the HCT116 cells.
Hyperthermia exposure upregulates
miR-34a and activates p53 transcriptional activity
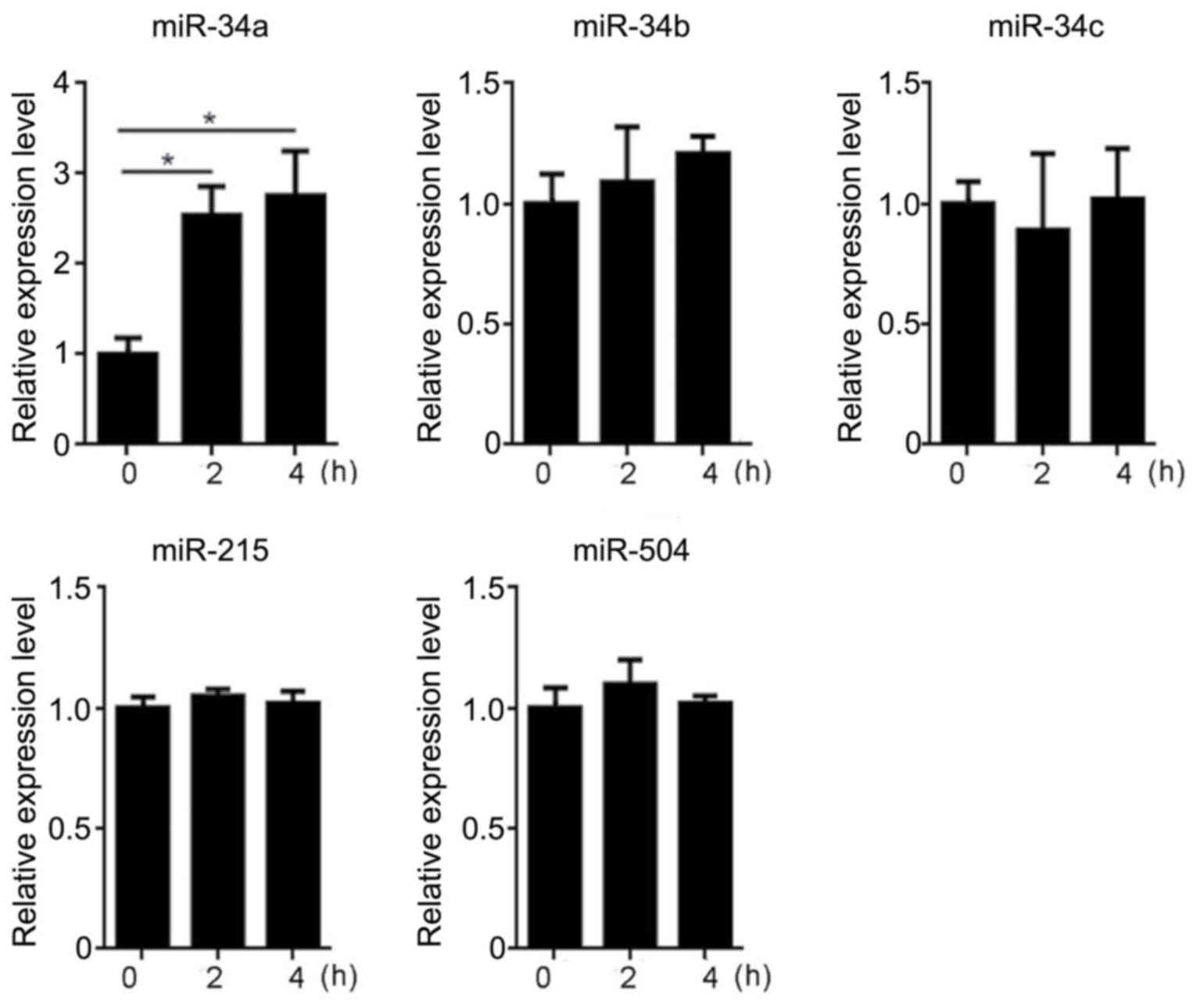
To investigate whether hyperthermia could affect the
expression of microRNAs potentially involved in a p53-miRNA
feedback loop, the HCT116 cells incubated at 37 or 42°C for 2 h
were subjected to RT-qPCR to evaluate the expression of miR-34a,
miR-34b, miR-34c, miR-215 and miR-504. The results of RT-qPCR
indicated that the levels of miR-34b, miR-34c, miR-215 and miR-504
did not differ significantly in the CTL or HYP groups over the 4 h
incubation period (data not shown). By contrast, significant
upregulation of miR-34a was identified in the HYP group after 2 and
4 h of hyperthermia (P<0.05; Fig.
2). Notably, compared with the CTL group, the expression of
miR-34a was increased by 2.5 or 2.9 fold after hyperthermia
exposure for 2 or 4 h, respectively, suggesting that miR-34a
expression was increased by hyperthermia.
To assess the effects of the hyperthermia-induced
miR-34a upregulation on p53 expression, RT-qPCR was performed to
evaluate the effect of miR-34a overexpression on the downstream
target genes transcriptionally activated by p53, including p21,
BAX, MDM2, PUMA and GADD45A (21).
Compared with the CTL group, hyperthermia exposure for 2 h
significantly promoted the expression of miR-34a (P<0.05), and
as expected, this upregulation in miR-34a caused significant
increases in the expression of all target genes assessed
(P<0.05; Fig. 3A). To determine
whether this increased gene expression was dependent on the
presence of the p53 protein, the PCR assay was repeated in
HCT116TP53-/− cells following hyperthermia exposure.
Similar to the HCT116 cells, the HCT116TP53-/− cells
exhibited a significant upregulation in miR-34a following
hypothermia exposure (P<0.05). However, hyperthermia exposure
failed to affect the expression of p21, BAX, MDM2 and GADD45A in
the HCT116TP53-/− cells (Fig.
3B). Interestingly, upregulated PUMA mRNA was still observed in
the HCT116TP53-/− cells, as in the HCT116 cells,
following hypothermia exposure (P<0.05). Luciferase reporter
assays were subsequently performed in the HCT116 cells to assess
whether hyperthermia-induced miR-34a expression enhanced the
promoter activity of the p53 consensus binding sequence within a
pGL3-P53BS vector. As depicted in Fig.
3C, hyperthermia exposure increased luciferase activity
compared with CTL group over time (P<0.05 vs. 0 h HYP). The
transfection of miR-34a and miR-34b by 3.5 and 2.7 fold,
respectively, significantly increased luciferase activity, which
confirmed the induction of luciferase activity by overexpression of
miR-34a and miR-34b (Fig. 3D,
P<0.05 vs. Scrambled mimics). No significant difference was
observed in luciferase activity following transfection with miR-34b
or miR-34c mimics.
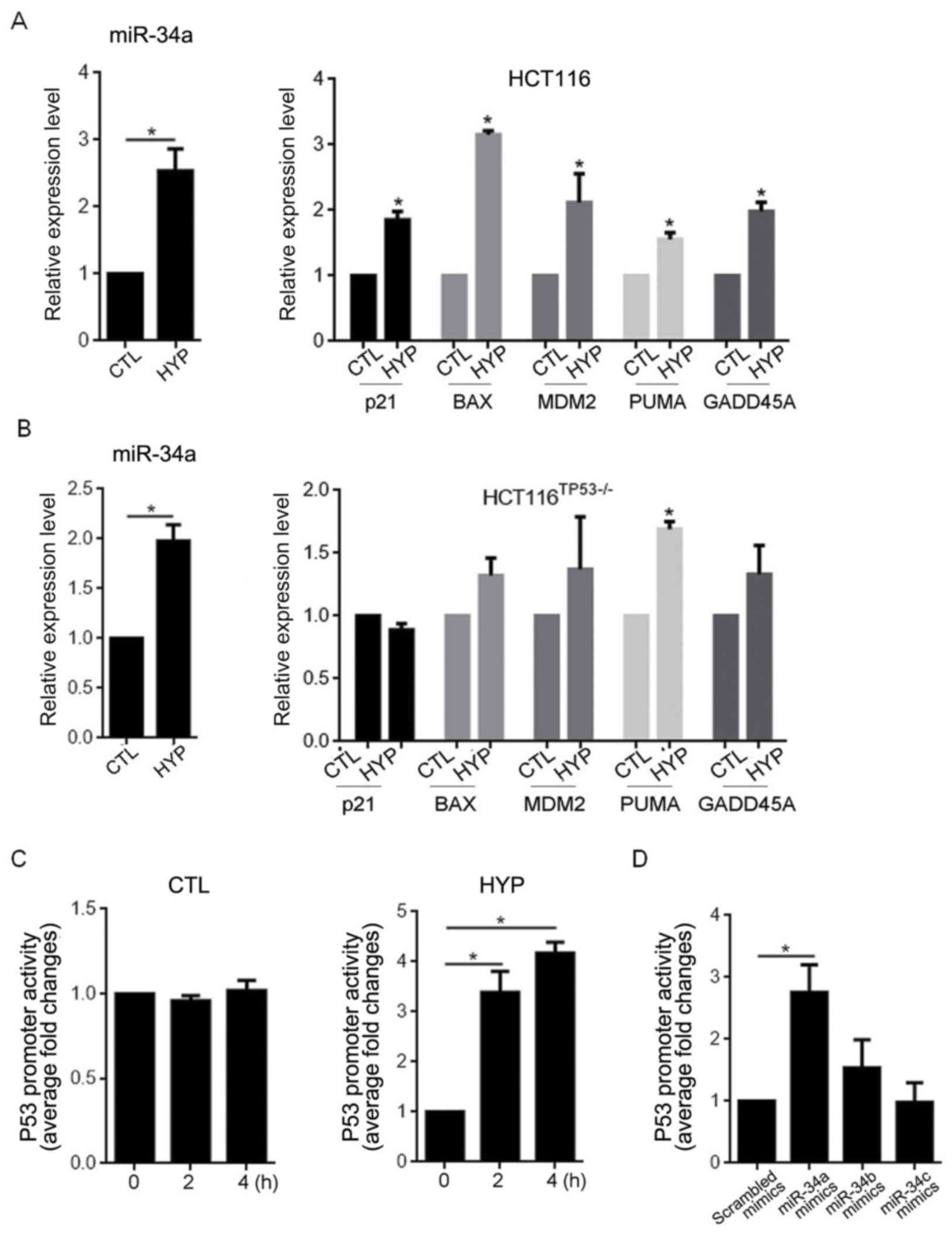 | Figure 3.Hyperthermia-induced miR-34a
expression increases p53 transcriptional activity. Reverse
transcription-quantitative polymerase chain reaction was performed
to measure the expression of miR-34a, p21, BAX, MDM2, PUMA and
GADD45A in (A) HCT116 and (B) HCT116TP53-/− cells
following hyperthermia exposure. (C) A luciferase reporter assay
was performed to assess the transcriptional activity of p53
following hyperthermia exposure. (D) Detection of p53
transcriptional activity following miR-34a transfection.
*P<0.05. miR, microRNA; BAX, B cell lymphoma 2-associated X
protein; MDM2, mouse double minute 2 homolog; PUMA, p53 upregulated
modulator of apoptosis; growth arrest and DNA-damage-inducible 45α;
HYP, hyperthermia; CTL, control. |
Hyperthermia promotes apoptosis
partially through upregulating miR-34a in HCT116 cells
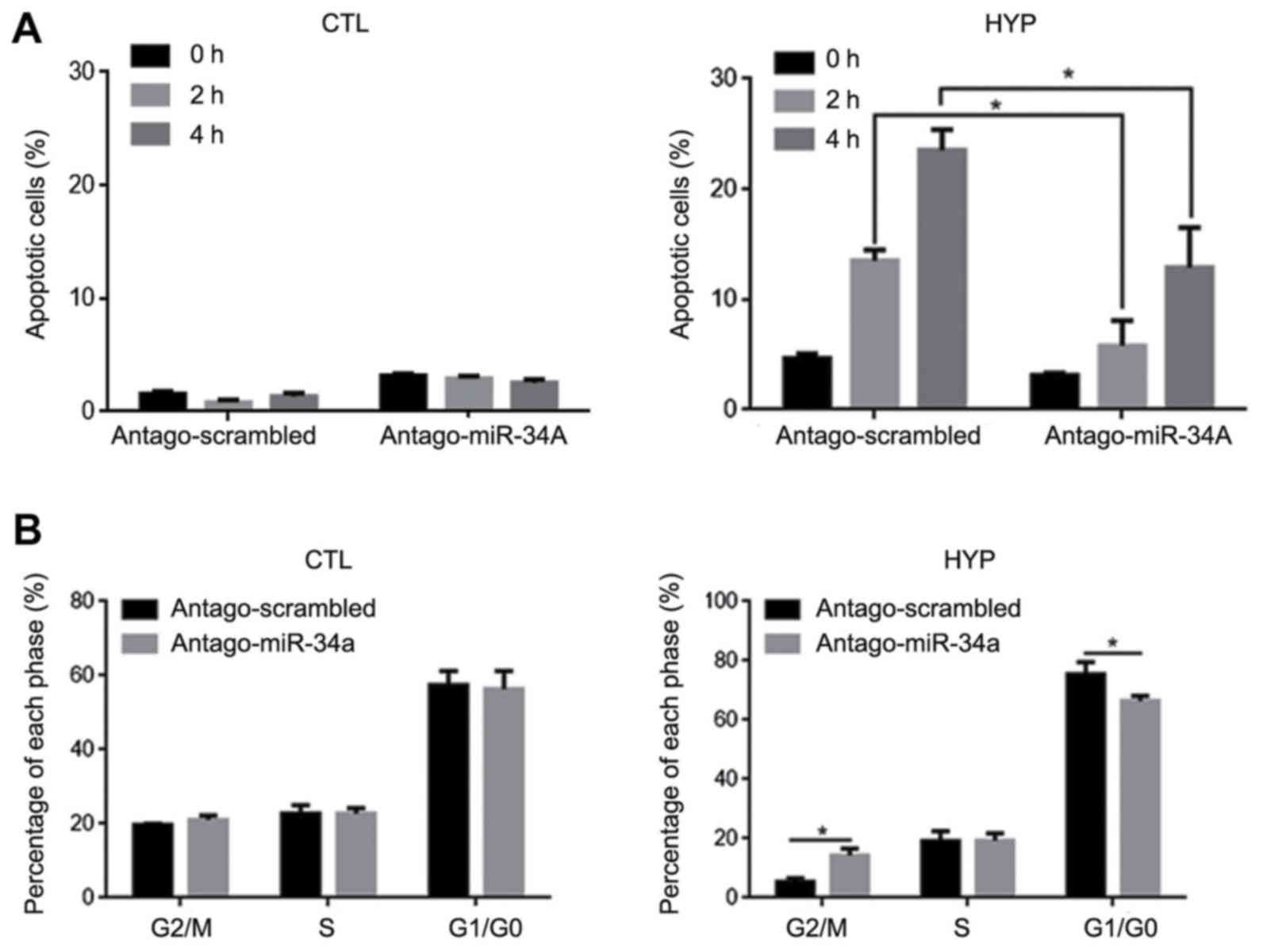
To determine whether hyperthermia induced apoptosis
and cell cycle arrest in HCT116 cells by stimulating miR-34a
expression, antago-miR-34a was transfected into HCT116 cells prior
to hyperthermia exposure. At 24 h posttransfection, the cells were
exposed to hyperthermia. The results indicated that, in the HCT116
cells, the introduction of antago-miR-34a decreased the elevated
rate of apoptosis induced by hyperthermia exposure after 2 and 4 h,
relative to the antago-scrambled group at the same time points
(P<0.05; Fig. 4A). Upon
determining the effect on cell proliferation, as expected, the
introduction of antago-miR-34a decreased the percentage of
G1/G0-phase cells and increased the percentage of G2/M-phase cells
following hyperthermia exposure for 4 h (P<0.05 vs.
antago-scrambled group; Fig.
4B).
Collectively, the present results indicate that
hyperthermia exposure enhances apoptosis and cell cycle arrest at
least in part through the upregulation of miR-34a, which may thus
activate the transcriptional activity of p53 in HCT116 cells;
however, the exact mechanisms underlying the upregulation of
miR-34a remain to be elucidated.
Discussion
In addition to surgery, chemotherapy, radiation and
biological therapy, hyperthermia has become a leading strategy for
the treatment of cancer, and is a key part of multidisciplinary
therapy (9). In RCC, hyperthermia is
accepted as an alternative method to surgery in certain patients
who are not suitable as surgical candidates (22). In CRC, hyperthermia is also used as
an alternative method in selected patients (23). However, hyperthermia presents a
variable success as an anti-cancer therapy in CRC. While neoplastic
cells exhibit relatively higher sensitivity to hyperthermia than
normal cells, multiple factors cause the variable outcomes due to
the cancer cells also exhibiting differential degrees of
sensitivity to hyperthermia (24,25).
Bordonaro et al (23)
reported that, in CRC, the mutation status of Kirsten rat sarcoma
viral oncogene homolog (KRAS) determined cell sensitivity to
hyperthermia, as cells lacking functional KRAS exhibited sustained
hyperactivation of extracellular signal-regulated kinase signaling
and increased Wingless/integrated-t-catenin signaling. It has also
been observed that mutations in TP53 increase the resistance of
cancer cells to hyperthermia by inactivating the downstream
regulated genes of p53 (26,27). Therefore, understanding the
mechanisms of hyperthermia-induced apoptosis and cell cycle arrest
in CRC cells may elucidate methods of promoting the sensitivity of
CRC to hyperthermia.
p53 is a transcription factor that modulates the
expression of numerous downstream target genes responsible for
directly and/or indirectly controlling apoptosis, cell cycle
arrest, senescence, DNA repair and genetic stability in response to
various cellular stresses (28),
including hyperthermia (29). It has
been reported that hyperthermia exposure promotes apoptosis
depending on the activation status of p53 (26). This indicates an association of
hyperthermia-induced apoptosis and cell cycle arrest with the
downstream gene targets of p53, which serve critical roles in cell
cycle arrest (p21, GADD45A), apoptotic cell death (PUMA, BAX), and
p53 feedback regulation (MDM2) (21).
In the present study, hyperthermia increased the
expression of miR-34a, but not of its family members miR-34b and
miR-34c. It is established that miR-34a serves anti-proliferative
and apoptotic roles by contributing to p53 function, principally
through targeting multiple inhibitors of p53, including proteins
that deacetylate p53 (sirtuin 1, metastasis associated 1 family
member 2/histone deacetylase 1) (30–33) and
the negative transcriptional modulator MDM4 (34). The present data demonstrated that
miR-34a expression was induced by hyperthermia exposure
independently of p53, thus indicating a link in a p53 feedback
loop. The results also suggested that upregulated miR-34a
expression following hyperthermia exposure may have increased the
sensitivity of the HCT116 cells to hyperthermia treatment.
Surprisingly, overexpression of miR-34a in HCT116 cells slightly
increased apoptotic sub-population, indicates the potential
pro-apoptotic role of miR-34a. Notably, the G1/G0 phase arrest
caused by hyperthermia exposure or the introduction of miR-34a
indicated a potential mechanism by which hyperthermia induced
apoptosis and inhibited proliferation.
In conclusion, the present data demonstrate for the
first time that hyperthermia may upregulate the expression of
miR-34a which may thus cause the transcriptional activation of p53
to induce apoptosis and cell cycle arrest in G1/G0 phase in HCT116
cells. These results provide novel insight into hyperthermia
cytotoxicity, and may indicate therapeutic targets regarding
hyperthermia sensitivity in CRC.
Acknowledgements
The present study was supported by the Scientific
and Technical Supporting Project of Wanzhou District, Chongqing
(grant no. 201403007).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Winawer SJ, Zauber AG, O'Brien MJ, Ho MN,
Gottlieb L, Sternberg SS, Waye JD, Bond J, Schapiro M, Stewart ET,
et al: Randomized comparison of surveillance intervals after
colonoscopic removal of newly diagnosed adenomatous polyps. The
National Polyp Study Workgroup. N Engl J Med. 328:901–906. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kornmann M, Formentini A, Ette C,
Henne-Bruns D, Kron M, Sander S, Baumann W, Kreuser ED, Staib L and
Link KH: Prognostic factors influencing the survival of patients
with colon cancer receiving adjuvant 5-FU treatment. Eur J Surg
Oncol. 34:1316–1221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zlobec I and Lugli A: Prognostic and
predictive factors in colorectal cancer. J Clin Pathol. 61:561–569.
2008.PubMed/NCBI
|
|
5
|
Oei AL, Vriend LE, Crezee J, Franken NA
and Krawczyk PM: Effects of hyperthermia on DNA repair pathways:
One treatment to inhibit them all. Radiat Oncol. 10:1652015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fałkowska-Podstawka M and Wernicki A: Heat
shock proteins in health and disease. Pol J Vet Sci. 6:61–70.
2003.PubMed/NCBI
|
|
7
|
Kim SY, Lee DH, Song X, Bartlett DL, Kwon
YT and Lee YJ: Role of Bcl-xL/Beclin-1 in synergistic apoptotic
effects of secretory TRAIL-armed adenovirus in combination with
mitomycin C and hyperthermia on colon cancer cells. Apoptosis.
19:1603–1615. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kalamida D, Karagounis IV, Mitrakas A,
Kalamida S, Giatromanolaki A and Koukourakis MI: Fever-range
hyperthermia vs. Hypothermia effect on cancer cell viability,
proliferation and HSP90 expression. PLoS One. 10:e01160212015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qi D, Hu Y, Li J, Peng T, Su J, He Y and
Ji W: Hyperthermia induces apoptosis of 786-O cells through
suppressing Ku80 expression. PLoS One. 10:e01229772015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wilmink GJ, Roth CL, Ibey BL, Ketchum N,
Bernhard J, Cerna CZ and Roach WP: Identification of microRNAs
associated with hyperthermia-induced cellular stress response. Cell
Stress Chaperones. 15:1027–1038. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu J, Liu F, Yin P, Zhu X, Cheng G, Wang
N, Lu A, Luan W, Zhang N, Li J, et al: Integrating miRNA and mRNA
expression profiles in response to heat stress-induced injury in
rat small intestine. Funct Integr Genomics. 11:203–213. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oshlag JZ, Devasthanam AS and Tomasi TB:
Mild hyperthermia enhances the expression and induces oscillations
in the Dicer protein. Int J Hyperthermia. 29:51–61. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Olivier M, Eeles R, Hollstein M, Khan MA,
Harris CC and Hainaut P: The IARC TP53 database: New online
mutation analysis and recommendations to users. Hum Mutat.
19:607–614. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Efeyan A and Serrano M: p53: Guardian of
the genome and policeman of the oncogenes. Cell Cycle. 6:1006–1010.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Graeber TG, Osmanian C, Jacks T, Housman
DE, Koch CJ, Lowe SW and Giaccia AJ: Hypoxia-mediated selection of
cells with diminished apoptotic potential in solid tumours. Nature.
379:88–91. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ren YL, Yu JH and Kinghorn AD: Development
of anticancer agents from Plant-derived Sesquiterpene lactones.
Curr Med Chem. 23:2397–2420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Navarro F and Lieberman J: miR-34 and p53:
New insights into a complex functional relationship. PLoS One.
10:e01327672015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yamakuchi M, Ferlito M and Lowenstein CJ:
miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci
USA. 105:pp. 13421–13426. 2008, View Article : Google Scholar : PubMed/NCBI
|
|
19
|
el-Deiry WS, Tokino T, Velculescu VE, Levy
DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and
Vogelstein B: WAF1, a potential mediator of p53 tumor suppression.
Cell. 75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sullivan KD, Gallant-Behm CL, Henry RE,
Fraikin JL and Espinosa JM: The p53 circuit board. Biochim Biophys
Acta. 1825:229–244. 2012.PubMed/NCBI
|
|
22
|
Campbell SC, Novick AC, Belldegrun A,
Blute ML, Chow GK, Derweesh IH, Faraday MM, Kaouk JH, Leveillee RJ,
Matin SF, et al: Guide lines for management of the clinical T1
renal mass. J Urol. 182:1271–1279. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bordonaro M, Shirasawa S and Lazarova DL:
In Hyperthermia Increased ERK and WNT signaling suppress colorectal
cancer cell growth. Cancer (Basel). 8:pii:E492016. View Article : Google Scholar
|
|
24
|
Hobohm U, Stanford JL and Grange JM:
Pathogen-associated molecular pattern in cancer immunotherapy. Crit
Rev Immunol. 28:95–107. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Trieb K, Sztankay A, Amberger A, Lechner H
and Grubeck-Loebenstein B: Hyperthermia inhibits proliferation and
stimulates the expression of differentiation markers in cultured
thyroid carcinoma cells. Cancer Lett. 87:65–71. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guan J, Stavridi E, Leeper DB and Iliakis
G: Effects of hyperthermia on p53 protein expression and activity.
J Cell Physiol. 190:365–374. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Okamoto K, Shinoura N, Egawa N, Asai A,
Kirino T, Shibasaki F and Shitara N: Adenovirus-mediated transfer
of p53 augments hyperthermia-induced apoptosis in U251 glioma
cells. Int J Radiat Oncol Biol Phys. 50:525–531. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li T, Kon N, Jiang L, Tan M, Ludwig T,
Zhao Y, Baer R and Gu W: Tumor suppression in the absence of
p53-mediated cell cycle arrest, apoptosis, and senescence. Cell.
149:1269–1283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gu ZT, Wang H, Li L, Liu YS, Deng XB, Huo
SF, Yuan FF, Liu ZF, Tong HS and Su L: Heat stress induces
apoptosis through transcription-independent p53-mediated
mitochondrial pathways in human umbilical vein endothelial cell.
Sci Rep. 4:44692014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Juan LJ, Shia WJ, Chen MH, Yang WM, Seto
E, Lin YS and Wu CW: Histone deacetylases specifically
down-regulate p53-dependent gene activation. J Biol Chem.
275:20436–20443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo J, Nikolaev AY, Imai S, Chen D, Su F,
Shiloh A, Guarente L and Gu W: Negative control of p53 by Sir2alpha
promotes cell survival under stress. Cell. 107:137–148. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Luo J, Su F, Chen D, Shiloh A and Gu W:
Deacetylation of p53 modulates its effect on cell growth and
apoptosis. Nature. 408:377–381. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vaziri H, Dessain SK, Eaton Ng E, Imai SI,
Frye RA, Pandita TK, Guarente L and Weinberg RA: hSIR2(SIRT1)
functions as an NAD-dependent p53 deacetylase. Cell. 107:149–159.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Finch RA, Donoviel DB, Potter D, Shi M,
Fan A, Freed DD, Wang CY, Zambrowicz BP, Ramirez-Solis R, Sands AT
and Zhang N: Mdmx is a negative regulator of p53 activity in vivo.
Cancer Res. 62:3221–3225. 2002.PubMed/NCBI
|