Introduction
Thrombotic thrombocytopenic purpura (TTP) is a rare
thrombotic microangiopathy (TMA) that may cause microangiopathic
hemolytic anemia, thrombocytopenia, fever, central nervous system
(CNS) symptoms and renal injury, which have been described as the
TTP pentad (1). TTP has a high
incidence in adults, is more common in females than males, and
occurs after infection, autoimmune disease, pregnancy, medication,
hematopoietic stem cell transplantation or drug allergy. The
incidence of TTP in countries outside China is 3.7/1,000,000, and
there is no definite statistical data in China (2). With in-depth understanding of this
disease, the diagnosis rate has increased. There is an increased
number of patients with TTP occurring following medication or other
diseases, with the incidence of 2/1,000,000 to 8/1,000,000
(3). Prior to the introduction of
plasma exchange (PE) treatment, patients with TTP typically had a
poor prognosis; however, unlike other types of TMA, TTP may now be
treated effectively by PE (4). The
mortality rate of TTP has dropped from 95 to 20% since the
introduction of PE treatment, but early diagnosis is critical to
ensure the most optimal possible outcome for patients (4). The occurrence of TTP is associated with
vascular endothelial cell injury, plasma von Willebrand factor
(vWF) and vWF cleaving protease (vWF-cp). Infection, autoimmune
diseases and/or medication cause vascular endothelial cell damage,
release of a large number of vWF, lack of vWF-cp or inhibition of
vWF-cp activity, leading to microaggregation of platelets and
vWF-fibrinogen, vessel occlusion, and rapid reduction of platelets,
and finally resulting in occurrence of TTP (5). The present case report described a
patient with TTP, who presented to the China-Japan Union Hospital
of Jilin University (Changchun, China), and the changes in vWF and
vWF-cp observed during treatment.
Case report
The present study was approved by the Ethics
Committee of China-Japan Union Hospital of Jilin University and
informed, written consent was received from the patient in the
present study. A 47-year-old man was admitted in February 2014 to
the China-Japan Union Hospital of Jilin University complaining of a
19-day history of fever, and a 2-h history of right limb
dysfunction and loss of speech. The patient's peak body temperature
was 37.7°C, and the patient had reported a small amount of gingival
bleeding. The sudden onset of neurologic symptoms included
difficulty standing, walking and holding items due to right-sided
weakness, slow response to commands and inability to speak. His
right limb symptoms had improved by the time of hospital admission
but his other symptoms had not. The patient had a 2-year history of
arterial hypertension, with the highest recorded blood pressure
(BP) of 160/90 mmHg; however, he had not received treatment with
antihypertensive agents.
Clinical examination on admission revealed a BP of
110/80 mmHg (normal range, 80–120/60-80 mmHg) and dysarthria;
cognitive function, including problem solving and memory, was
impaired, MMSE 17 points (university education level, ≤23 points
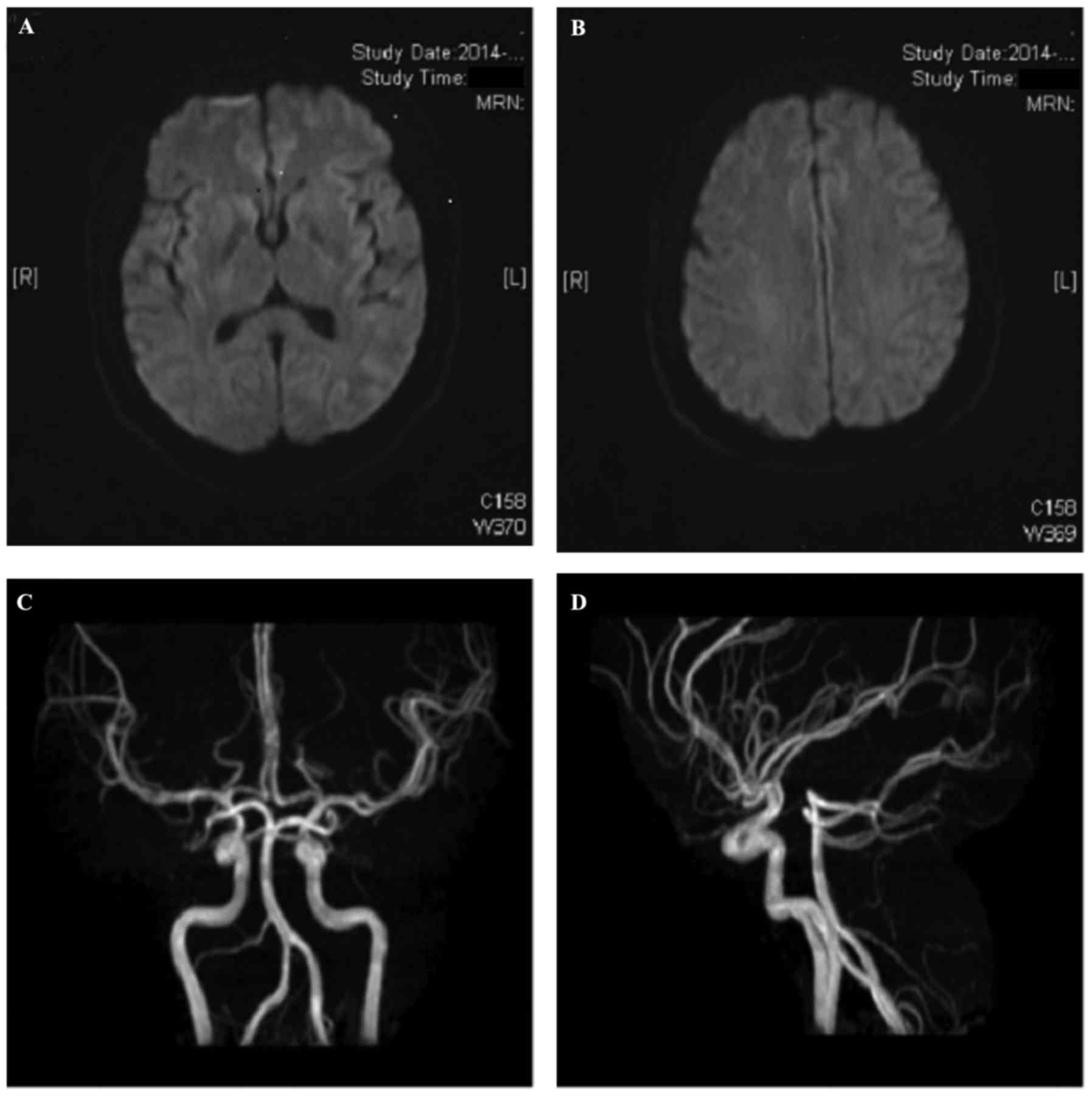
indicative of cognitive dysfunction) (6). Computed tomography and
diffusion-weighted magnetic resonance (MR) imaging of the brain,
and cerebral MR angiography were performed, but no abnormalities
were identified (Fig. 1). Laboratory
investigations indicated a white blood cell (WBC) count of
12.8×109 cells/l (reference range, 4–10×109
cells/l), a red blood cell (RBC) count of 2.5×1012
cells/l (reference range, 3.5–5.5×1012 cells/l), a
hemoglobin (Hb) concentration of 75.0 g/l (reference range, 110–160
g/l) and platelet (PLT) count of 11×109 cells/l
(reference range, 100–300×109 cells/l). Urinalysis
revealed a WBC count of 107.1 cells/µl (reference range, 0–25
cells/µl) and a RBC of 84.2 cells/µl (reference range, 0–25
cells/µl). Dipstick urinalysis detected protein 2+ (Neg), occult
blood 3+ (Neg), urobilinogen 1+ (Neg) and urobilin 1+ (Neg). Liver
function tests revealed serum concentrations of alanine
transaminase of 323 IU/l (reference range, 5–40 IU/l), aspartate
aminotransferase of 114 IU/l (reference range, 8–40 IU/l),
γ-glutamyl transferase of 72 IU/l (reference range, 8–57 IU/l) and
glutamate dehydrogenase of 35 U/l (reference range, 0–10 U/l), a
total bilirubin concentration of 46.0 µmol/l (reference range, 5–21
µmol/l), a direct bilirubin concentration of 11.5 µmol/l (reference
range, 0–3 µmol/l) and an indirect bilirubin concentration of 34.5
µmol/l (reference range, 2–21 µmol/l). Renal function tests
revealed serum concentrations of urea of 12.4 µmol/l (reference
range, 3.2–7.1 µmol/l) and creatinine (SCr) of 126.7 µmol/l
(reference range, 58–110 µmol/l). A peripheral blood film (5) indicated mature erythrocytes of a
variety of sizes with RBC debris and scattered microspherocytes.
The reticulocyte count was 0.391×1012 cells/l (5), which accounted for 22% of the total
RBCs. Bone marrow examination suggested aplastic anemia and
thrombocytopenia (5). Antinuclear
antibodies were not detected (5).
Based on these findings, a diagnosis of TTP and grade 2
hypertension (1999 WHO/ISH Guidelines for the Management of
Hypertension) (7) (very high-risk
group) were determined.
A total of 4 days following admission, the patient
underwent PE for the first time: cell harvesting was performed
using Cobe Spectra cell separator (Terumo BCT, Inc., Lakewood, CO,
USA). Frozen plasma fluid (2,000–3,000 ml) was replaced once a day
for 13 days. Plasma was replaced at 15–20 ml/min and blood flow
velocity was 120–150 ml/min. After low-dose low molecular weight
heparin anticoagulation, 30 min after plasma replacement, 10%
calcium gluconate (10 ml/h) was intravenously administered.
Glucocorticoids (Pfizer, Inc., New York, NY, USA) were administered
(1.0 g once daily for 3 days, 0.5 g once daily for 3 days, 0.24 g
once daily for 3 days, 0.12 g once daily for 3 days, 0.08 g once
daily for 3 days and 0.04 g once daily for 3 days, all via
intravenous drip) and supportive care was given (roton pump
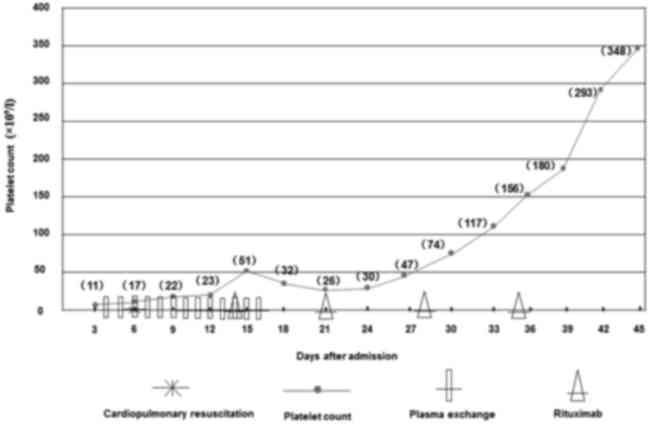
inhibitor, potassium, fat milk and compound amino acids). As shown
in Fig. 2, patients were
administered PE therapy starting from 4 days after admission, once
a day, for a total of 13 days. A total of 2 days after PE
administration, the patient sustained a cardiac arrest and required
closed-chest cardiac massage and cardiopulmonary resuscitation, and
an autonomous cardiac rhythm and respiration were restored. A total
of 14 days following admission, a regime of 100 mg rituximab (Roche
Diagnostics, Basel, Switzerland) infused intravenously once a week
for 4 weeks was initiated. By the time of his discharge 6 weeks
after admission, the patient had undergone 13 sessions of PE
treatment. The patient was asymptomatic and exhibited no abnormal
CNS signs. The complete blood count before the patient was
discharged indicated a WBC count of 5.3×109 cells/l, a
RBC count of 3.3×1012 cells/l, a Hb concentration of
115.0 g/l and a PLT count of 348×109 cells/l. Urinalysis
and liver function tests were normal. Monthly rituximab was
administered for 4 times after discharge to prevent relapse, and no
recurrence was detected at the 20-month follow-up after
discharge.
A 4-ml specimen of the morning fasting venous blood
was obtained from the patient at different phases of disease (at 8,
10, 23 and 32 days after admission) to allow comparison with two
healthy volunteers. These two healthy volunteers (a 33-year-old
male and a 35-year-old male) were persons who received physical
examination in January 2016 in the outpatient clinic of China-Japan
Union Hospital of Jilin University. The volunteers signed informed
consent regarding participation in this study. Blood was taken in
EDTA tubes and centrifuged at 1,500 × g, 4°C for 20 min.
Supernatant plasma was collected and stored at −20°C prior to
measurement of vWF and vWF-cp concentrations. Measurements were
performed using a double antibody sandwich ELISA kit (CSB-E08437h;
Cusabio Biotech Co., Ltd., Wuhan, China) according to the
manufacturer's protocols.
Following PE, hematologic and biochemical parameters
(analyzed using an automatic biochemical analyzer) began to
gradually improve over the 32 days after admission; PLT, SCr and
serum lactate dehydrogenase concentration returned to their normal
ranges. Furthermore, plasma vWF concentrations decreased and vWF-cp
activity increased (Table I). On day
8 after admission, the patient's vWF concentration was increased
compared with the normal control, then gradually decreased, and was
close to normal level at discharge. On day 8 after admission, the
patient's vWF-cp activity was markedly increased compared with the
normal control, maintained at a very high level, and it did not
recover to the normal level at discharge.
 | Table I.Comparison of the patient's
hematologic profile with healthy controls. |
Table I.
Comparison of the patient's
hematologic profile with healthy controls.
|
| Patient results
according to hospital stay, days |
|
|
|---|
|
|
|
|
|
|---|
| Variable | 8 | 10 | 23 | 32 | Control | Control |
|---|
| Complete blood
count |
|
|
|
|
|
|
|
Platelets, ×109
cells/l | 41 | 51 | 62 | 193 | – | – |
| White
blood cells, ×109 cells/l | 25 | 15 | 8 | 4 | – | – |
|
Hemoglobin concentration,
g/l | 99 | 89 | 86 | 108 | – | – |
| Number of PE
sessions | 6 | 10 | 12 | 12 | – | – |
| Number of rituximab
treatments | 0 | 0 | 2 | 4 | – | – |
| Serum creatinine
concentration, µm/ml | 153.5 | 134.7 | 89 | 85 | – | – |
| Lactate
dehydrogenase, U/l | 1,770 | 1,131 | 343 | 265 | – | – |
| vWF, ng/ml | 12.80 | 16.59 | 13.75 | 8.59 | 4.72 | 5.82 |
| vWF-cleaving
protease, U/ml | 36.39 | 36.30 | 58.89 | 50.89 | 8.10 | 3.23 |
Once PLT had returned to the normal range
(193×109 cells/l) at 32 days after admission, the
patient's plasma vWF concentration was broadly comparable with that
of healthy volunteers. Additionally, plasma vWF-cp activity had
markedly increased compared with that of healthy volunteers
controls (Table I). Plasma vWF
concentration decreased to normal levels over the 32 days after
admission, and vWF-cp activity remained very high and did not
recover to normal level at discharge (Table I).
Discussion
TTP is rare, rapidly progressive and it has very
high mortality rate if plasma replacement therapy is not given
(8). TTP has been indicated to cause
thrombocytopenia and thrombogenesis in the small arteries and
capillaries (9). The mortality rate
of TTP is 90% without PE, however, the introduction of PE has
improved the mortality rate (4). A
study by Bukowski et al (10)
first reported the successful treatment of TTP with PE in 1976, and
PE has now become the first choice treatment for TTP. Timely,
extensive PE has been indicated to reduce the mortality rate to
<10%, resulting in >90% short-term effectiveness (4). PE should be sustained for 2–3 days
after the restoration of PLT to 150×109 cells/l, after
which it may be stopped (11).
However, a longer course of treatment may be required for patients
at risk of long-term relapse who respond poorly to PE (12). Although a standard dose (375 mg
rituximab/m2 body surface area administered once a week
for 4 consecutive weeks) of rituximab may also be effective for
these patients, the high cost of this treatment limits its use
(13). Rituximab is a biosynthesized
mouse/human chimeric monoclonal antibody that specifically binds
cluster of differentiation 20 antigens on the B-lymphocyte surface,
preferentially clearing those abnormally reacting with autoantigens
by inducing complement-dependent cytotoxicity and
antibody-dependent cell-mediated cytotoxic reactions (14). Rituximab targets and selectively
removes abnormal B-lymphocytes, and has therefore been widely used
for the treatment of autoimmune diseases in which B-lymphocytes are
dysfunctional (14). For TTP
treatment, rituximab removes the lymphocytes responsible for
producing vWF (14). It has been
reported that 375 mg rituximab per square meter body surface area
administered once a week for 4 consecutive weeks is a highly
effective treatment for idiopathic TTP, particularly for patients
with low vWF-cp activity and vWF-cp-positive antibodies, and has
been indicated to result in a remission rate of ~95% and a
recurrence rate of ~10% (14). In
patients that experience TTP recurrence, further use of rituximab
may improve the patient outcome (15). Given that the cost of rituximab is
high, and when used to treat TTP it is administered outside its
licensed indications (rituximab is typically used to treat
B-lymphocyte-mediated diseases, including non-Hodgkin's lymphoma,
chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis
with polyangiitis and microscopic polyangiitis), it has been
suggested that low-dose rituximab is a satisfactory treatment for
TTP (16). In the present case
study, continued administration of low-dose rituximab after PE was
provided to the patient and no relapse was indicated at the
20-month follow-up after discharge.
A study by Moake et al (17) first detected vWF in the serum of
patients with TTP in 1982, laying the foundations for the study of
TTP pathogenesis. In 1996, a study by Furlan et al (18) isolated a serum metalloproteinase that
cleaves vWF, and while the cause of TTP is not known, it was found
that patients with TTP lack this metalloproteinase (18). In 2001, a study by Gerritsen et
al (19) purified the
metalloproteinase and demonstrated that it belonged to an ADAMTS
gene family, the genes for vWF-cp (ADAMTS13) have been mapped to
chromosome 9q34 (19). Previous
findings have recognized that deletion of ADAMTS13 may be
acquired or hereditary, but both result in sustained vWF-dependent
aggregation of platelets, microthrombus formation and TTP (20). These findings suggest that TTP may be
caused by relative or absolute deficiencies in the activity of
plasma vWF-cp. For most patients with hereditary TTP, the vWF-cp
activity is always <5%, and may be 0% in those with idiopathic
TTP (21). A decrease in activity to
<5% may indicate the formation of plasma anti-vWF-cp antibodies
(21). Previous results suggest that
prognosis appears favorable if vWF-cp activity rises to ~50% and
the antibody titer falls after PE (18). In addition, although not all patients
with TTP present with severe depletion of vWF-cp activity, it may
still help guide treatment (22).
Among TTP survivors who have a vWF-cp activity <10%, nearly half
relapse, most commonly in the second year after the acute event
(22). Conversely, those with normal
vWF-cp activity after treatment are unlikely to suffer from
recurrence (22). This suggests that
any factor causing vWF-cp depletion at any time may increase the
risk of recurrence for patients who have recovered from TTP. Some
results have concluded that vWF-cp activity >10% of the normal
range identifies patients at high risk of recurrence (23). In the present case, vWF concentration
and vWF-cp activity was measured at 8, 10, 23 and 32 days after
admission; when compared with the controls, results suggested that
the patient's vWF concentration gradually decreased and vWF-cp
activity slowly increased to 51%, which was suggestive of a
favorable prognosis and a lower risk of recurrence.
The patient's vWF concentration and vWF-cp activity
was compared with controls after the patient's PLT had been
restored at day 32 after admission. Results indicated that the vWF
concentration was broadly comparable with the controls, but were
marginally higher in the patient, which was a likely cause of the
elevated vWF-cp activity in the patient in comparison with the
healthy volunteers, which is consistent with previous findings
(24). However, vWF-cp concentration
was lower than expected given the findings of previous studies.
Further studies in larger cohorts are required to fully illuminate
vWF-cp activity in TTP. According to the current findings, PE is
the preferred treatment for TTP and it can markedly decrease the
mortality rate of TTP. A standard or low dose of rituximab is
effective for treatment of TTP and lowers the recurrence rate. The
occurrence of TTP is closely related to vWF and vWF-cp.
Acknowledgements
The present study was supported by the Finance
Department of Jilin Province in China (grant no. 3D514L533430).
References
|
1
|
Amorosi EL and Ultmann JE: Thrombotic
thrombocytopenic purpura: Report of 16 cases and review of the
literature. Medicine. 45:139–159. 1966. View Article : Google Scholar
|
|
2
|
Allford SL, Hunt BJ, Rose P and Machin SJ:
Haemostasis and Thrombosis Task Force, British Committee for
Standards in Haematology: Guidelines on the diagnosis and
management of the thrombotic microangiopathic haemolytic anaemias.
Br J Haematol. 120:556–573. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Said A, Haddad RY, Stein R and Lerma EV:
Thrombotic thrombocytopenic purpura. Dis Mon. 60:500–504. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Edel E, Al-Ali HK, Seeger S, Kauschat D
and Matthes G: efficacy and safety profile of solvent/detergent
plasma in the treatment of acute thrombotic thrombocytopenic
purpura: A single-center experience. Transfus Med Hemother.
37:13–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ye YF, Wang YS and Shen ZY: National Guide
to Clinical Laboratory Procedures. Third. Southeast University
Press; Nanjing: 2006
|
|
6
|
Rovner BW and Folstein MF: Mini-mental
state exam in clinical practice. Hosp Pract (Off Ed).
22:991031061101987.PubMed/NCBI
|
|
7
|
Chalmers J: The 1999 WHO-ISH guidelines
for the management of hypertension. Med J Aust. 171:458–459.
1999.PubMed/NCBI
|
|
8
|
Bell WR, Braine HG, Ness PM and Kickler
TS: Improved survival in thrombotic thrombocytopenic
purpura-hemolytic uremic syndrome. Clinical experience in 108
patients. N Engl J Med. 325:398–403. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
George JN: How I treat patients with
thrombotic thrombocytopenic purpura: 2010. Blood. 116:4060–4069.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bukowski RM, Hewlett JS, Harris JW,
Hoffman GC, Battle JD Jr, Silverblatt E and Yang IY: Exchange
transfusions in the treatment of thrombotic thrombocytopenic
purpura. Semin Hematol. 13:219–232. 1976.PubMed/NCBI
|
|
11
|
Fontana S, Hovinga JA Kremer, Lämmle B and
Taleghani B Mansouri: Treatment of thrombotic thrombocytopenic
purpura. Vox Sang. 90:245–254. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rizzo C, Rizzo S, Scirè E, Di Bona D,
Ingrassia C, Franco G, Bono R, Quintini G and Caruso C: Thrombotic
thrombocytopenic purpura: A review of the literature in the light
of our experience with plasma exchange. Blood Transfus. 10:521–532.
2012.PubMed/NCBI
|
|
13
|
Westwood JP, Webster H, McGuckin S,
McDonald V, Machin SJ and Scully M: Rituximab for thrombotic
thrombocytopenic purpura: Benefit of early administration during
acute episodes and use of prophylaxis to prevent relapse. J Thromb
Haemost. 11:481–490. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Heidel F, Lipka DB, von Auer C, Huber C,
Scharrer I and Hess G: Addition of rituximab to standard therapy
improves response rate and progression-free survival in relapsed or
refractory thrombotic thrombocytopenic purpura and autoimmune
haemolytic anaemia. Thromb Haemost. 97:228–233. 2007.PubMed/NCBI
|
|
15
|
Foley SR, Webert K, Arnold DM, Rock GA,
Clark WF, Barth D and Sutton DM; Members of the Canada Apheresis
Group (CAG), : A Canadian phase II study evaluating the efficacy of
rituximab in the management of patients with relapsed/refractory
thrombotic thrombocytopenic purpura. Kidney Int Suppl. S55–S58.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tun NM and Villani GM: Efficacy of
rituximab in acute refractory or chronic relapsing non-familial
idiopathic thrombotic thrombocytopenic purpura: A systematic review
with pooled data analysis. J Thromb Thrombolysis. 34:347–359. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moake JL, Rudy CK, Troll JH, Weinstein MJ,
Colannino NM, Azocar J, Seder RH, Hong SL and Deykin D: Unusually
large plasma factor VIII: Von willebrand factor multimers in
chronic relapsing thrombotic thrombocytopenic purpura. N Engl J
Med. 307:1432–1435. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Furlan M, Robles R and Lämmle B: Partial
purification and characterization of a protease from human plasma
cleaving von willebrand factor to fragments produced by in vivo
proteolysis. Blood. 87:4223–4234. 1996.PubMed/NCBI
|
|
19
|
Gerritsen HE, Robles R, Lämmle B and
Furlan M: Partial amino acid sequence of purified von willebrand
factor-cleaving protease. Blood. 98:1654–1661. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hovinga JA Kremer and Lämmle B: Role of
ADAMTS13 in the pathogenesis, diagnosis, and treatment of
thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ
Program. 2012:610–616. 2012.PubMed/NCBI
|
|
21
|
Duraković N, Radonić R and Gasparović V:
Thrombotic thrombocytopenic purpura-the role of ADAMTS13 assay in
clinical practice. Coll Antropol. 34:1087–1091. 2010.PubMed/NCBI
|
|
22
|
Hovinga JA Kremer, Vesely SK, Terrell DR,
Lämmle B and George JN: Survival and relapse in patients with
thrombotic thrombocytopenic purpura. Blood. 115:1500–1511. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peyvandi F, Lavoretano S, Palla R, Feys
HB, Vanhoorelbeke K, Battaglioli T, Valsecchi C, Canciani MT,
Fabris F, Zver S, et al: ADAMTS13 and anti-ADAMTS13 antibodies as
markers for recurrence of acquired thrombotic thrombocytopenic
purpura during remission. Haematologica. 93:232–239. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arya M, Anvari B, Romo GM, Cruz MA, Dong
JF, McIntire LV, Moake JL and López JA: Ultralarge multimers of von
willebrand factor form spontaneous high-strength bonds with the
platelet glycoprotein Ib-IX complex: Studies using optical
tweezers. Blood. 99:3971–3977. 2002. View Article : Google Scholar : PubMed/NCBI
|