Introduction
Prostate cancer (PCa), as one of the most common
men's malignancy in America, is the second leading cause of
cancer-related death in men (1).
Although more than 80% of PCa was diagnosed as localized disease
and commonly treated by radical prostatectomy (RP), postoperative
recurrence occurred in about 15% of patients within 5 years and up
to 40% within 10 years (2).
Recurrence of localized PCa following treatment can lead to lethal
metastatic castration-resistant PCa. Various biomarkers have been
reported for PCa recurrence surveillance, including preoperative
prostate specific antigen (PSA) value, Gleason score, lymph node
invasion and others, but not cancer-specific and inaccurate
(3). Therefore, more efforts should
be devoted for identifying disease specific markers of PCa
recurrence that can better directly offer practical aid to drug
treatment and lead to improved survival and reductions in
morbidity.
Although the mechanism underlying PCa is not yet
completely understood, multiple genes to help predict PCa risk have
been proposed by considerable researches. Brian R. Hu et al
(4) reported that AXIN2 expression
could not only predict PCa recurrence, but also promoted tumor
growth and metastasis in vivo and vitro. Hao et
al (5) found that XPO6
expression was elevated in PCa and maybe a potential prognostic
biomarker for PCa recurrence. Additionally, some other targets from
blood and (or) urine have been examined and identified, including
KLK2-KLK3 SNP rs2735839, 17p12 SNP rs4054823 and Eotaxin-1
(6,7). However, few of these profiles have been
adopted in the clinic after RP to predict recurrence PCa.
Therefore, there is still a need for novel tumor biomarkers that
can help improve prediction of prostate cancer recurrence upon
clinical variables.
To explore more meaningful molecular biomarker for
predicting the prostate cancer prognosis, technologies with
high-throughput screen was implied to identify the genes.
Microarray data GSE 25136 with 39 recurrent and 40 non-recurrent
PCa was published and analyzed by Stephenson et al (8) via leave-one-out-cross-validation
(LOOCV) approach and the results showed that Etoposide-induced 2.4
mRNA (EI24) and mitogen-activated protein kinase kinase kinase
kinase 4 (MAP4K4) were the most highly overexpressed genes and
erythrocyte membrane protein band 4.9 (EPB49) was the most highly
underexpressed gene in recurrent tumors compared with primary PCa
and may be the potential biomarker. Subsequently, Sun and Goodison
(9) conducted a more advanced
computational algorithm to analyze the Microarray data GSE25136 and
acquire more accurate biomarkers for predicting the prognosis of
PCa. With technological development, bioinformatics has been a
mainstream tool to analyze the microarray data. In the present
study, microarray data GSE25136 (8,9)
was employed to identify differentially expressed genes (DEGs)
between PCa and PCa recurrence samples with Limma package in R
language. Furthermore, gene ontology (GO) and pathway enrichment
analysis was performed to screen the DEGs. Lastly, PPI networks of
DEGs was constructed by Cytosacpe mapping software and hub genes
was identified by the STRING database. Therefore, it is better for
us to further understand the molecular mechanisms of PCa.
Materials and methods
Microarray data
The gene expression profiles of GSE25136 were
downloaded from the GEO database. GSE25136 based on Affymetrix
GPL96 platform (Affymetrix Human Genome U133A Array), was submitted
by Sun and Goodison (9) and updated
on Jul 01, 2016. The GSE25136 dataset contained 79 PCa samples
treated by radical prostatectomy (RP) in 1993 and 1999, including
39 recurrent and 40 non-recurrent PCa samples. When serum level of
PSA consecutively increased at least 3 times post operation, the
patients were classified as disease recurrence; non-recurrent
patients with an undetectable PSA (<0.05 ng/ml) for at least 5
years after RP were identified. The clinical characteristics of all
79 patients has been completely described by Stephenson et
al (8). In briefly, Median PSA
level and Prostatectomy Gleason sum of patients with recurrence
were higher than those in non-recurrenct PCa patients, and the
number of patients with Extracapsular extension, positive surgical
margins, seminal vesicle invasion were greater in recurrent PCa
group compared with non-recurrent PCa group.
Identification of DEGs
The raw data files used for the analysis included
cell files (Affymertix platform). The data was preprocessed by R
biocondutor RMA Packages, and DEGs were identified by limma
packages in recurrent PCa compared with non-recurrent PCa samples.
DEGs were identified with a change fold and defined a P-value
cutoff of <0.05 to be statistically significant. Hierarchical
clustering analysis was applied to categorize the data into two
groups that had similar expression patterns. Heatmap was performed
by the pheatmap package analysis with joint between-within
distances. Expression values from multiple clones or probe sets
mapping to the same Unigene Cluster ID were averaged.
Gene ontology (GO) analysis of
DEGs
The Database for Annotation, Visualization and
Integrated Discovery (DAVID; https://david.ncifcrf.gov/) provides a comprehensive
set of novel and powerful tools for assigning biological meaning to
a set of genes (10). The false
recovery rate (FDR) <0.05 was used as the cut-off criterion for
GO functional enrichment analysis by DAVID.
Pathway enrichment analysis of DEGs in
the regulatory network
KEGG (http://www.genome.jp/) is acknowledge base for
systematic analysis of gene functions, linking genomic information
with higher-order functional information (11). P<0.05 was used as the cut-off
criterion for the Kyoto Encyclopedia of Genes and Genomes pathway
enrichment analysis using DAVID.
Integration of protein-protein
interaction (PPI) network and module analysis
The search Tool for the Retrieval of Interacting
Genes (STRING) database is an online tool designed to evaluate PPI
information. STRING (version 10.0) covers 5,214,234 proteins from
1,133 organisms. To evaluate the interactive relationships between
the DEGs, the DEGs were mapped to STRING, and only experimentally
validated interactions with a combined score >0.4 were selected
as significant. Then, the Cytoscape software was used to construct
the PPI network. The plug-in Molecular Complex Detection (MCODE)
was used to screen the modules of PPI network in Cytoscape with a
degree cutoff=2, node score cutoff=0.2, k-core=2, max depth from
seed=100. The criteria were set as follows: MCODE scores >4 and
number of nodes >4. P<0.05 was considered to have significant
differences.
Results
Identification of DEGs
After data, including 39 recurrent PCa samples and
40 non-recurrent PCa samples, was downloaded from GEO database and
preprocessed, 708 DEGs, including 212 up genes and 496 down genes
were identified using limma packages on the basis of the cut-off
criteria (P<0.05 and fold control (FC) ≥1.4 criteria) in
recurrent samples compared with non-recurrent samples.
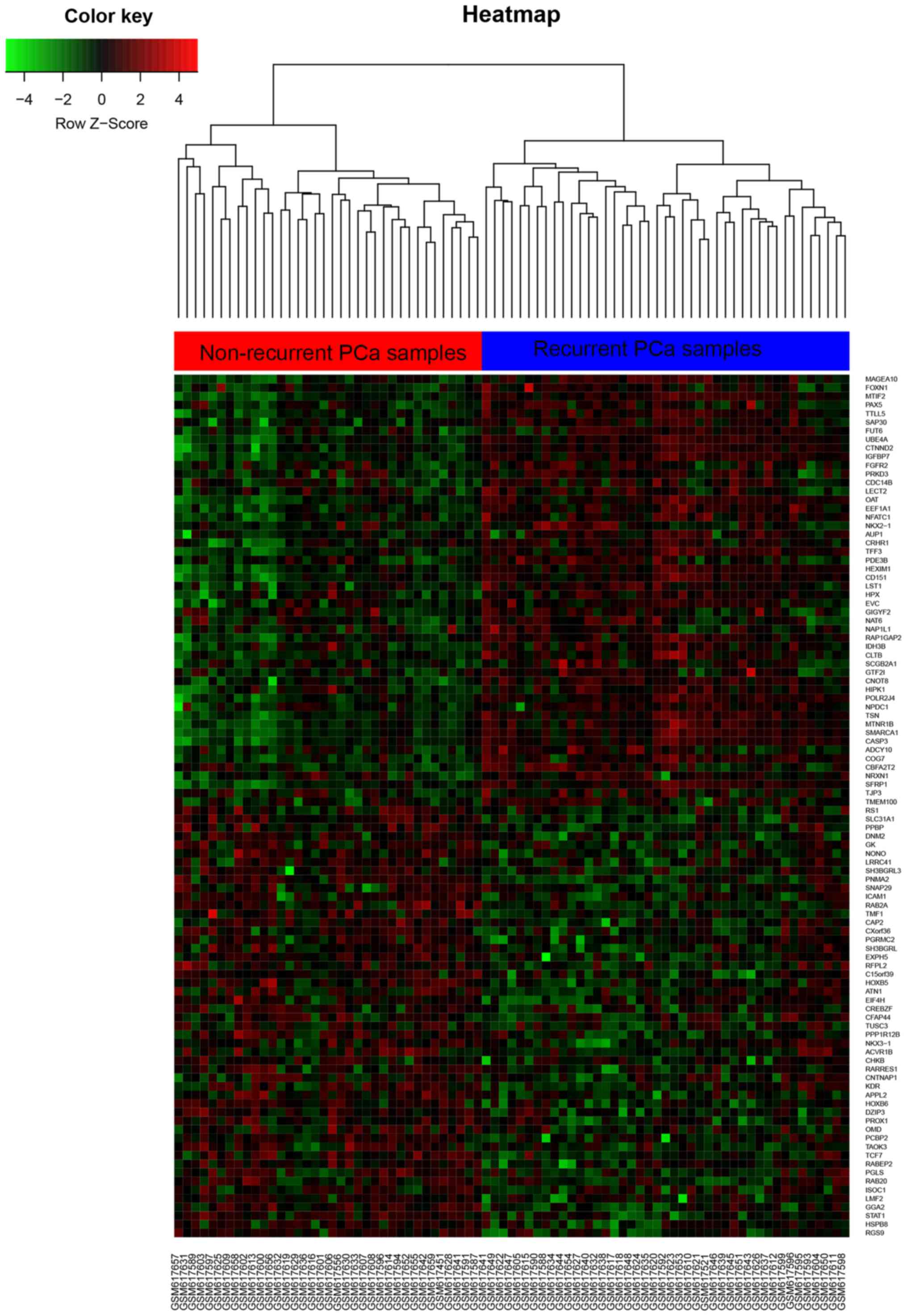
Subsequently, DEGs were performed hierarchical clustering analysis
and it can accurately classify the prostate samples as recurrent
PCa tissues and non-recurrent PCa tissues (Fig. 1: left, recurrent PCa; right,
non-recurrent PCa). Additionally, Top up 50 DEGs and down 50 DEGs
expression with most significant was shown in Fig. 1 (P<0.05).
GO functional enrichment analysis
In order to gain further insight into the function
of the identified DEGs, we uploaded DEGs to the online biological
classification software DAVID to identify typical GO categories. Go
analysis showed that DEGs were significantly enriched in biological
processes (BP), including cell adhesion, negative regulation of
growth, extracellular matrix organization, negative regulation of
cell migration, apoptotic signaling pathway (Table I). For cell components, DEGs were
enriched in focal adhesion, extracellular exosome, cell-cell
adherens junction, proteinaceous extracellular matrix (Table I). Finally, Go molecular function
analysis showed that DEGs were enriched in protein binding, protein
homodimerization activity, cadherin binding involved in cell-cell
adhesion, insulin receptor binding (Table I).
 | Table I.Gene ontology analysis of
differentially expressed genes associated with PCa recurrence. |
Table I.
Gene ontology analysis of
differentially expressed genes associated with PCa recurrence.
| Category | Term/gene
function | Gene count | % | P-value |
|---|
| GOTERM_BP_DIRECT | GO:0007155~cell
adhesion | 38 | 6.551724138 | 3.82E-07 |
| GOTERM_BP_DIRECT | GO:0045926~negative
regulation of growth |
8 | 1.379310345 | 1.33E-06 |
| GOTERM_BP_DIRECT |
GO:0030198~extracellular matrix
organization | 22 | 3.793103448 | 1.72E-06 |
| GOTERM_BP_DIRECT | GO:0030336~negative
regulation of cell migration | 12 | 2.06896552 | 2.57E-04 |
| GOTERM_BP_DIRECT | GO:0097190~apoptotic
signaling pathway | 10 | 1.72413793 | 4.71E-04 |
| GOTERM_CC_DIRECT | GO:0005925~focal
adhesion | 42 | 7.241379 | 8.83E-12 |
| GOTERM_CC_DIRECT |
GO:0070062~extracellular exosome | 149 | 25.68966 | 1.26E-11 |
| GOTERM_CC_DIRECT | GO:0005913~cell-cell
adherens junction | 23 | 3.965517241 | 4.87E-04 |
| GOTERM_CC_DIRECT |
GO:0005578~proteinaceous extracellular
matrix | 23 | 3.965517 | 3.33E-05 |
| GOTERM_MF_DIRECT | GO:0005515~protein
binding | 372 | 64.13793103 | 3.67E-15 |
| GOTERM_MF_DIRECT | GO:0042803~protein
homodimerization activity | 46 | 7.931034483 | 2.34E-05 |
|
GOTERM_MF_DIRECT | GO:0098641~cadherin
binding involved in cell-cell adhesion | 20 | 3.448275862 | 0.002756127 |
|
GOTERM_MF_DIRECT | GO:0005158~insulin
receptor binding |
6 | 1.034482759 | 0.0028733 |
KEGG pathway analysis
We employed KEGG pathway enrichment analysis to
identify the most significantly enriched pathways of the DEGs. 10
biological pathways which significant enriched with DEGs including
cAMP signaling pathway, MAPK signaling pathway, Adherensjunction,
Calcium signaling pathway, Pathways in cancer, Proteoglycans in
cancer, Transcriptional misregulation in cancer, Leukocyte
transendothelial migration, Focal adhesion and Ras signaling
pathway (Table II).
| Table II.KEGG pathway analysis of
differentially expressed genes associated with PCa recurrence. |
Table II.
KEGG pathway analysis of
differentially expressed genes associated with PCa recurrence.
| Pathway ID | Name | Gene count | % | P-value | Genes |
|---|
| hsa04024 | cAMP signaling
pathway | 23 | 0.021929197 | 5.74E-05 |
PLD1,VAV3,PTGER3,PDE3B, GRIA3, PDE4D,
GABBR2, CNGB1, BDNF, HTR1A,ATP2B4, NPY, GRIA2, PDE4A, RAC1,
CREB3L2, RYR2, GNAS, CAMK2B, ADCY10, FSHB, GLP1R, NFATC1 |
| hsa04010 | MAPK signaling
pathway | 22 | 0.020975754 | 0.004317491 | FGFR2, FGF8, FGF7,
CACNA1I, TAOK3, MAPK11, MECOM, FLNC, FLNB, CDC42, MAP4K4, CASP3,
BDNF, RPS6KA4, PAK2, SOS1, RAC1, CACNA1G, EGF, DUSP7, MAP2K5,
NFATC1 |
| hsa04520 | Adherens
junction | 9 | 0.00858099 | 0.012675942 | ACTB, CDC42, TCF7,
CSNK2A1, BAIAP2, RAC1, SSX2IP, PTPN1, ACTN3 |
| hsa04020 | Calcium signaling
pathway | 16 | 0.015255094 | 0.012798992 | SLC8A2, PTGER3,
SPHK2, SPHK1, CACNA1I, VDAC1, GNAL, ATP2B4, ATP2A3, RYR3, PDE1A,
CACNA1G, RYR2, GNAS, CAMK2B, ADRA1D |
| hsa05200 | Pathways in
cancer | 27 | 0.025742971 | 0.025195199 | FGFR2, FGF8, TCF7,
PTGER3, CTBP2, FGF7, RXRB, ITGA2, IGF1, STAT1, MECOM, CDC42, CASP3,
LAMB2, HDAC2, CXCR4, SOS1, RAC1, MDM2, NKX3-1, GNAS, GNB3, GNG3,
RARB, EGF, WNT6, APC |
| hsa05205 | Proteoglycans in
cancer | 16 | 0.015255094 | 0.031484838 | ACTB, PPP1R12B,
ITGA2, IGF1, MAPK11, FLNC, FLNB, KDR, CDC42, CASP3, HPSE, SOS1,
RAC1, MDM2, CAMK2B, WNT6 |
| hsa05202 | Transcriptional
misregulation in cancer | 14 | 0.013348207 | 0.035045912 | SUPT3H, FLT1, RXRB,
IGF1, PAX5, GRIA3, GZMB, HDAC2, REL, LYL1, MDM2, PBX1, IGFBP3,
CDK14 |
| hsa04670 | Leukocyte
transendothelial migration | 11 | 0.010487877 | 0.035804774 | ACTB, CDC42, ICAM1,
NOX3, VAV3, CXCR4, CLDN5, NOX1, RAC1, MAPK11, ACTN3 |
| hsa04510 | Focal adhesion | 16 | 0.015255094 | 0.039374107 | ACTB, CDC42, FLT1,
VAV3, LAMB2, PAK2, SOS1, PPP1R12B, RAC1, IGF1, ITGA2, ACTN3, FLNC,
EGF, FLNB, KDR |
| hsa04014 | Ras signaling
pathway | 17 | 0.016208537 | 0.042199384 | FGFR2, PLD1, FGF8,
FGF7, FLT1, IGF1, BRAP, KDR, CDC42, PAK2, REL, SOS1, TEK, RAC1,
GNG3, GNB3, EGF |
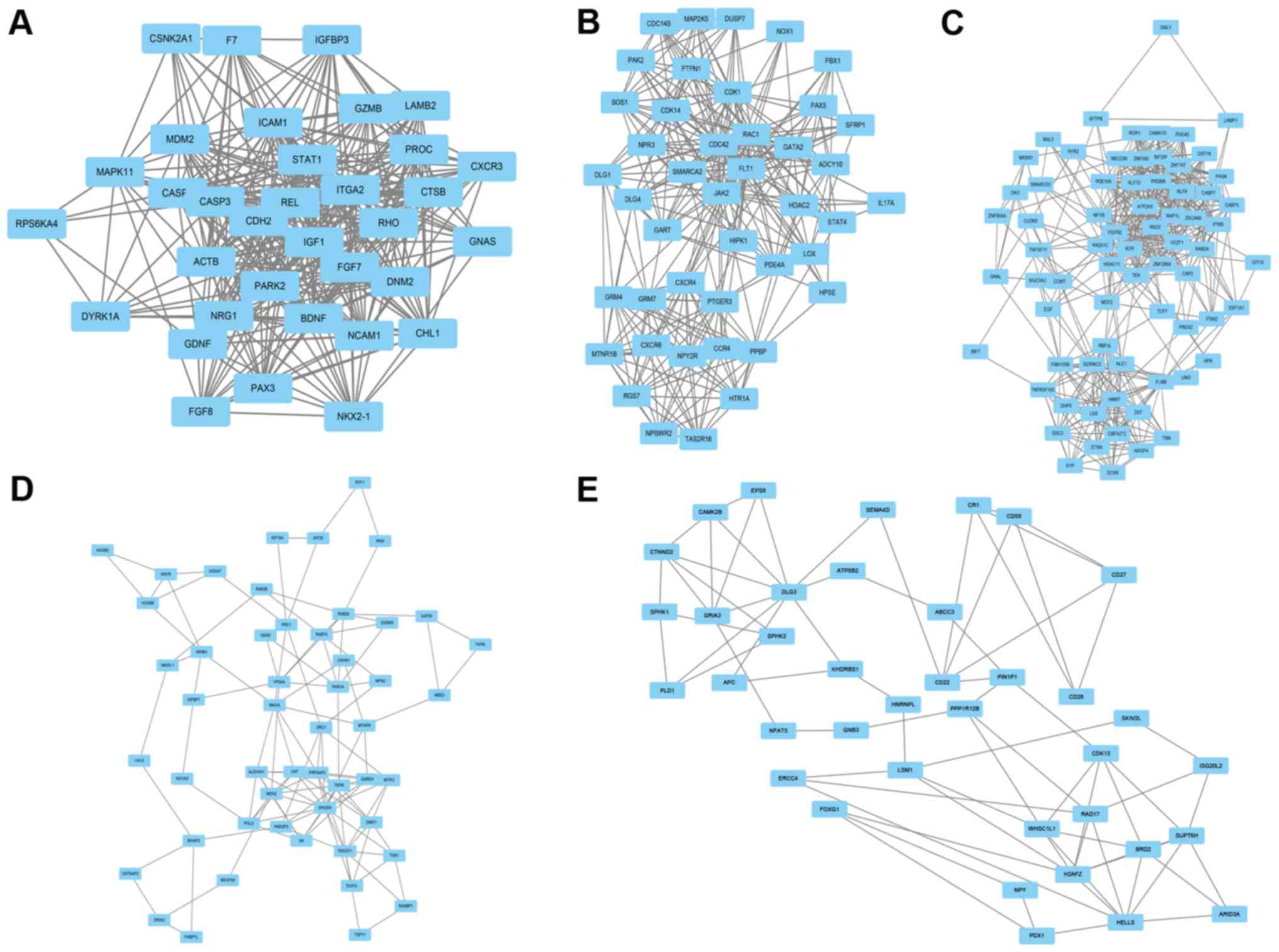
Construction of the PPI network
Cytosacpe mapping software was employed to construct
the PPI network of DEGs. A total of 663 nodes and 8,871 edges were
analyzed using plug-ins MCODE. The top 5 significant modules with
MCODE scores >4 and nodes >4 in whole network were screened
by analysis in the STRING database, and the hub gene in each
cluster, also called the seed, was identified by on the basis of
the highest modules scoring in the cluster including Insulin-like
growth factor-1 (IGF-1) (Fig. 2A),
mitogen-activated protein kinase kinase 5 (MAP2K5) (Fig. 2B), Receptor tyrosine kinase like
orphan receptor 1 (ROR1) (Fig. 2C),
Inhibin beta A (INHBA) (Fig. 2D),
and differentiation-22 (CD22) (Fig.
2E). All clusters are named after the hub gene name, and of
these clusters, IGF-1 modules showed a highest MCODE scores in
whole network, with 20.606. Additionally, IGF-1 modules consisted
of 34 nodes and 340 edges; MAP2K5 modules consisted of 43 nodes and
306 edges; ROR1 modules consisted of 71 nodes and 464 edges; INHBA
modules consisted of 51 nodes and 119 edges; CD22 modules consisted
of 38 nodes and 74 edges.
Discussion
PCa is the fourth leading global cause of human
malignancies worldwide, and is a product of mutation in genomics
including cumulative genetic, epigenetic, somatic, and endocrine
aberrations (12). Of the
differential expression of genes caused by various mutations, some
specific genes are positively or negatively associated with therapy
resistance and poor outcomes in PCa. The wide application of
microarray and high through put sequencing has made it possible to
identify the more appropriate genes to predict the prognosis of PCa
after RP from thousands of genes in human genome level (12). In the present study, we extracted the
data from GSE25136 and 708 DEGs between recurrent PCa samples and
non-recurrent PCa samples using bioinformatics analysis were
screened out. Functional annotation showed that these DEGs were
mainly involved in cell adhesion, focal adhesion, protein binding,
cAMP signaling pathway and MAPK signaling pathway. In addition, to
better understand the interaction of these DEGs, a PPI network was
constructed and we identified four key genes, including CD22,
IGF-1, INHBA, MAP2K5 and ROR1, that can provide new ideas for
predict the prognosis in PCa following RP.
The GO term analysis showed that these DEGs were
mainly involved cell adhesion, focal adhesion, and protein binding.
In addition, cAMP and MAPK signaling pathway were shown to
participate int PCa recurrence by KEGG pathway analysis. Classical
signal transduction pathway and cAMP signaling pathway have been
extensively studied in the context of carcinogenesis by regulating
cellular growth and proliferation. cAMP-dependent protein kinase
(PKA), as a critical mediator of cAMP signaling pathway, has been
demonstrated that it is overexpressed in PCa and has been examined
as a potential biomarker for predicting the outcome of PCa patients
(13). Androgens are required for
the initiation and the development of PCa via stimulating the AR
signaling pathway, and androgen ablation therapies, such as
chemical or surgical castration, have become a standard against PCa
(14). There is a highly relevant
cross-talk between cAMP and AR signaling pathway in PCa
progression, because not only cAMP and PKA activation may result in
the stimulation of AR but androgens can also regulate the activity
of PKA (15). In addition, cyclic
nucleotide phosphodiesterases (PDEs) are involved in the metabolism
of cAMP by regulating its degradation (16). PDE4D, as a kind of PDEs, is highly
expressed in PCa and has been implicated to promote PCa progression
(16). Furthermore, members of the
PDE4D subfamily are classified as long, short and super-short.
PDE4D7, as a long isoform member, is downregulated in
androgen-independent prostate cancer cells compared with androgen
sensitive prostate cancer cells, and inhibited its growth by
compartmentalising cAMP (17). R
Böttcher et al (18) also
showed that it was up-regulated in localized PCa samples compared
with the normal adjacent prostate tissues, while its expression
diminished with emergence of Castration resistant prostate cancer
(CRPC). Other PDE4D isoform composition, such as PDE4D5 and PDE4D9,
was also upregulated in PCa and played an import role in PCa
progression (19). MAPK signaling
pathway also played a vital role in regulating cellular behaviors
in response to extracellular stimuli. Dysregulation of p38 MAPK, as
a main subgroup of MAPK signaling pathway, are associated with
tumor stages and poor survival of PCa patients (20). The emergence of Castration resistant
prostate cancer (CRPC) caused by certain co-activators or through
MAPK signaling pathway activities, which lead to the overexpression
of anti-apoptotic genes and survival of the cancer cells, thus
increasing the PCa related death (21).
Finally, the PPI network with DEGs was constructed
and the hub genes exhibiting the highest degree of connectivity
were identified, including CD22, IGF-1, INHBA, MAP2K5 and ROR1.
IGF-1 was identified as one of the most DEGs in the recurrent PCa
samples. IGF-1, also known as somatomedin 1, is a mitogen that
plays a key role in regulating various cell biological behavior,
including cell proliferation, differentiation, and apoptosis via
endocrine, paracrine and autocrine mechanisms (22). IGF-1 binds to the insulin-like growth
factor 1 recep-tor (IGF-1R), which is a tyrosine kinase receptor,
and initiates a cascade of downstream signal transduction pathways
(23). Results published recent
studies evaluated the role of IGF-1 in PCa and showed that higher
circulating IGF-1 levels were consistently associated with
increased risk of PCa in epidemio-logic studies (24). IGF-1, which is synthesized locally in
an autocrine or paracrine manner by PCa cells, may stimulate PCa
growth and development (25). Then,
IGF has been a pivotal target gene for PCa therapy. Magnolol has
been demonstrated that it served as a novel anti-PCa agenet via
regulating the expression of IGF-1 in vitro (26). Apigenin effectively suppressed PCa
cells growth and metastasis in TRAMP mice by attenuating IGF-I
signaling (27). In addition, IGF-1
genotypes and haplotypes were associated with wore survival of PCa
patients with bone metastasis (28).
Mitogen-activated protein kinase kinase 5 (MAP2K5), also known as
MEK5, was overexpressed in PCa, which was associated with tumor
metastases and unfavourable survival outcome of PCa patients
(29).
Receptor tyrosine kinase like orphan receptor 1
(ROR1), also known as neurotrophic tyrosine kinase,
receptor-related 1 (NTRKR1), is a transmembrane protein belonging
to the receptor tyrosine kinase (RTK) family. Down-regulation of
ROR1 inhibited human colorectal cancer cell growth and promoted
apoptosis (30). ROR1 has been shown
to be overexpressed in several solid tumors and its unique
expression by malignant cells surface is a target for novel
therapeutics, especially monoclonal antibodies (mAbs) for the
treatment of cancer (31). Inhibin
beta A, also known as INHBA, is a subunit of both activin and
inhibin, two closely related glycoproteins with opposing biological
effects. INHBA is overexpressed in various cancers, including
gastric cancer, rothelial carcinoma of the urinary bladder and
upper tract and colorectal cancer, demonstrating its association
with poor prognosis of patients (32–34).
Cluster of differentiation-22 (CD22), as a molecule belonging to
the SIGLEC family of lectins, is a transmembrane glycoprotein
expressed by mature B cells. Recently, Tuscano and colleagues
reported that CD22, a hallmark marker on B lymphocytes, was
expressed on lung cancer cells and might serve as a new target for
therapy (35). However, Pop et
al (36) reported that the
surface of lung cancer cells did not detect CD22 expression, and
cannot be killed by anti-CD22 immunotoxins, which have not
previously been directly associated with initiation and progression
of PCa, according to the present results.
Although data from GSE25136 have been analyzed by
previous authors, and some DEGs have been identified, the
uniqueness of the present study is that Limma package in R
language, as one of the most fashionable Algorithmic Language now,
was applied to analyze the data from GSE25136 and different DEGs
compared with previous report was identified. Furthermore, DEGs in
PCa recurrence related BP and signaling pathway was screened out,
which may help us better understand the potential mechanism of PCa
recurrence. Additionally, a PPI network of DEGs was constructed and
5 hub nodes with higher degrees were identified and could be used
to predict the prognosis of PCa.
In conclusion, the results of this study provide a
comprehensive bioinformatics analysis of DEGs to increase the
understading of the mechanism underlying PCa recurrence. The study
showed that CD22, IGF-1, INHBA, MAP2K5 and ROR1 may be pivotal for
participating in PCa recurrences. However, these functions need to
be confirmed by further molecular biological experiments.
References
|
1
|
Hu BR, Fairey AS, Madhav A, Yang D, Li M,
Groshen S, Stephens C, Kim PH, Virk N, Wang L, et al: AXIN2
expression predicts prostate cancer recurrence and regulates
invasion and tumor growth. Prostate. 76:597–608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li HY, Jin N, Han YP and Jin XF: Pathway
crosstalk analysis in prostate cancer based on protein-protein
network data. Neoplasma. 64:22–31. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stephenson AJ, Scardino PT, Eastham JA,
Bianco FJ Jr, Dotan ZA, DiBlasio CJ, Reuther A, Klein EA and Kattan
MW: Postoperative nomogram predicting the 10-year probability of
prostate cancer recurrence after radical prostatectomy. J Clin
Oncol. 23:7005–7012. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu BR, Fairey AS, Madhav A, Yang D, Li M,
Groshen S, Stephens C, Kim PH, Virk N, Wang L, et al: AXIN2
expression predicts prostate cancer recurrence and regulates
invasion and tumor growth. Prostate. 76:597–608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hao J, Chiang YT, Gout PW and Wang Y:
Elevated XPO6 expression as a potential prognostic biomarker for
prostate cancer recurrence. Front Biosci (Schol Ed). 8:44–55. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Choudhury AD, Eeles R, Freedland SJ,
Isaacs WB, Pomerantz MM, Schalken JA, Tammela TL and Visakorpi T:
The role of genetic markers in the management of prostate cancer.
Eur Urol. 62:577–587. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heidegger I, Höfer J, Luger M, Pichler R,
Klocker H, Horninger W, Steiner E, Jochberger S and Culig Z: Is
Eotaxin-1 a serum and urinary biomarker for prostate cancer
detection and recurrence? Prostate. 75:1904–1909. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stephenson AJ, Smith A, Kattan MW,
Satagopan J, Reuter VE, Scardino PT and Gerald WL: Integration of
gene expression profiling and clinical variables to predict
prostate carcinoma recurrence after radical prostatectomy. Cancer.
104:290–298. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun Y and Goodison S: Optimizing molecular
signatures for predicting prostate cancer recurrence. Prostate.
69:1119–1127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gene Ontology Consortium, . The Gene
Ontology (GO) project in 2006. Nucleic Acids Res. 34(Database
issue): D322–D326. 2006.PubMed/NCBI
|
|
11
|
Kanehisa M and Goto S.: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thoma C: Prostate cancer: The genomics of
localized disease. Nat Rev Urol. 14:652017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Khor LY, Bae K, Al-Saleem T, Hammond EH,
Grignon DJ, Sause WT, Pilepich MV, Okunieff PP, Sandler HM and
Pollack A: Protein kinase A RI-alpha predicts for prostate cancer
outcome: analysis of radiation therapy oncology group trial 86–10.
Int J Radiat Oncol Biol Phys. 71:1309–1315. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Govindan R: The Washington Manual of
Oncology. 2nd. Lippincott Williams and Wilkins; Philadelphia:
2008
|
|
15
|
Merkle D and Hoffmann R: Roles of cAMP and
cAMP-dependent protein kinase in the progression of prostate
cancer: Cross-talk with the androgen receptor. Cell Signal.
23:507–515. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rahrmann EP, Collier LS, Knutson TP, Doyal
ME, Kuslak SL, Green LE, Malinowski RL, Roethe L, Akagi K, Waknitz
M, et al: Identification of PDE4D as a proliferation promoting
factor in prostate cancer using a Sleeping Beauty transposon-based
somatic mutagenesis screen. Cancer Res. 69:4388–4397. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Henderson DJ, Byrne A, Dulla K, Jenster G,
Hoffmann R, Baillie GS and Houslay MD: The cAMP
phosphodiesterase-4D7 (PDE4D7) is downregulated in
androgen-independent prostate cancer cells and mediates
proliferation by compartmentalising cAMP at the plasma membrane of
VCaP prostate cancer cells. Br J Cancer. 110:1278–1287. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Böttcher R, Henderson DJ, Dulla K, van
Strijp D, Waanders LF, Tevz G, Lehman ML, Merkle D, van Leenders
GJ, Baillie GS, et al: Human phosphodiesterase 4D7 (PDE4D7)
expression is increased in TMPRSS2-ERG-positive primary prostate
cancer and independently adds to a reduced risk of post-surgical
disease progression. Br J Cancer. 113:1502–1511. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Böttcher R, Dulla K, van Strijp D, Dits N,
Verhoef EI, Baillie GS, van Leenders GJ, Houslay MD, Jenster G and
Hoffmann R: Human PDE4D isoform composition is deregulated in
primary prostate cancer and indicative for disease progression and
development of distant metastases. Oncotarget. 7:70669–70684.
2016.PubMed/NCBI
|
|
20
|
Koul HK, Pal M and Koul S: Role of p38 MAP
kinase signal transduction in solid tumors. Genes Cancer.
4:342–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guan M, Fousek K and Chow WA: Nelfinavir
inhibits regulated intramembrane proteolysis of sterol regulatory
element binding protein-1 and activating transcription factor 6 in
castration-resistant prostate cancer. FEBS J. 279:2399–2411. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bach LA and Hale LJ: Insulin-like growth
factors and kidney disease. Am J Kidney Dis. 65:327–336. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shanmugalingam T, Bosco C, Ridley AJ and
Van Hemelrijck M: Is there a role for IGF-1 in the development of
second primary cancers? Cancer Med. 5:3353–3367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cao Y, Nimptsch K, Shui IM, Platz EA, Wu
K, Pollak MN, Kenfield SA, Stampfer MJ and Giovannucci EL:
Prediagnostic plasma IGFBP-1, IGF-1 and risk of prostate cancer.
Int J Cancer. 136:2418–2426. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dunn SE, Kari FW, French J, Leininger JR,
Travlos G, Wilson R and Barrett JC: Dietary restriction reduces
insulin-like growth factor I levels, which modulates apoptosis,
cell proliferation, and tumor progression in p53-deficient mice.
Cancer Res. 57:4667–4672. 1997.PubMed/NCBI
|
|
26
|
McKeown BT and Hurta RA: Magnolol affects
expression of IGF-1 and associated binding proteins in human
prostate cancer cells in vitro. Anticancer Res. 34:6333–6338.
2014.PubMed/NCBI
|
|
27
|
Shukla S, MacLennan GT, Fu P and Gupta S:
Apigenin attenuates insulin-like growth factor-I signaling in an
autochthonous mouse prostate cancer model. Pharm Res. 29:1506–2617.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsuchiya N, Narita S, Inoue T, Saito M,
Numakura K, Huang M, Hatakeyama S, Satoh S, Saito S, Ohyama C, et
al: Insulin-like growth factor-1 genotypes and haplotypes influence
the survival of prostate cancer patients with bone metastasis at
initial diagnosis. BMC Cancer. 13:1502013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mehta PB, Jenkins BL, McCarthy L, Thilak
L, Robson CN, Neal DE and Leung HY: MEK5 overexpression is
associated with metastatic prostate cancer, and stimulates
proliferation, MMP-9 expression and invasion. Oncogene.
22:1381–1389. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma W, He X, Zhang H, Liu T, Feng X and
Zhou Q: Down-regulation of receptor-tyrosine-kinase-like orphan
receptor 1 suppresses cell growth and enhances apoptosis in human
colorectal carcinoma. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi.
32:655–665. 2016.(In Chinese). PubMed/NCBI
|
|
31
|
Hojjat-Farsangi M, Moshfegh A,
Daneshmanesh AH, Khan AS, Mikaelsson E, Osterborg A and Mellstedt
H: The receptor tyrosine kinase ROR1-an oncofetal antigen for
targeted cancer therapy. Semin Cancer Biol. 29:21–31. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oshima T, Yoshihara K, Aoyama T, Hasegawa
S, Sato T, Yamamoto N, Akito N, Shiozawa M, Yoshikawa T, Numata K,
et al: Relation of INHBA gene expression to outcomes in gastric
cancer after curative surgery. Anticancer Res. 34:2303–2309.
2014.PubMed/NCBI
|
|
33
|
Lee HY, Li CC, Huang CN, Li WM, Yeh HC, Ke
HL, Yang KF, Liang PI, Li CF and Wu WJ: INHBA overexpression
indicates poor prognosis in urothelial carcinoma of urinary bladder
and upper tract. J Surg Oncol. 111:414–422. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Okano M, Yamamoto H, Ohkuma H, Kano Y, Kim
H, Nishikawa S, Konno M, Kawamoto K, Haraguchi N, Takemasa I, et
al: Significance of INHBA expression in human colorectal cancer.
Oncol Rep. 30:2903–2908. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tuscano JM, Kato J, Pearson D, Xiong C,
Newell L, Ma Y, Gandara DR and O'Donnell RT: CD22 antigen is
broadly expressed on lung cancer cells and is a target for
antibody-based therapy. Cancer Res. 72:5556–5565. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pop LM, Barman S, Shao C, Poe JC, Venturi
GM, Shelton JM, Pop IV, Gerber DE, Girard L, Liu XY, et al: A
reevaluation of CD22 expression in human lung cancer. Cancer Res.
74:263–271. 2014. View Article : Google Scholar : PubMed/NCBI
|