Introduction
The sirtuin (Sirt) protein family belongs to the
class III of NAD-dependent histone deacetylases and comprises seven
members (termed Sirt1-7) (1). Sirt
proteins have been widely investigated for their deacetylation
activities, characteristic by deacetylating histones, such as H3,
and non-histone proteins, such as cluster of differentiation
(CD)K9; Sirt proteins are frequently overexpressed in several types
of cancer (2). Sirt7 is a histone H3
on lysine 18 (H3K18) deacetylase and, as a new member of this
family, has been reported to be mainly localized in the nucleus
(3). In addition, Sirt7 has been
reported by different researchers to be involved in certain
carcinomas, including ovarian (4),
gastric (5), breast (6) and cervical cancer (7). However, the role and function of Sirt7
in colorectal carcinoma (CRC) remains to be investigated. As Sirt1
serves a role in the promotion of epithelial-mesenchymal transition
and metastasis in colorectal cancer the function of Sirt7 in CRC
was explored.
CRC is the third most common malignancy worldwide
and the fourth leading cause of cancer-associated mortalities, with
rectal carcinoma constituting 28% of all CRC cases (8,9). Despite
the advances in surgery, chemotherapy and radiotherapy in the past
decades, various clinical side effects occur in these traditional
treatments (10,11). Furthermore, distant metastasis,
particularly liver metastasis, is the main cause of mortality in
patients with CRC, and the current therapy is largely unsuccessful
due to tumor resistance (12,13).
Therefore, improving the understanding of the tumorigenesis process
and the molecular mechanism underlying CRC is of great importance
for developing novel diagnostic and therapeutic strategies. The
present study aimed to explore the expression and function of Sirt7
in CRC, in order to bring novel insight into understanding the
mechanisms about CRC.
Materials and methods
Patients
A total of 60 fresh tumor tissues and their adjacent
non-tumorous tissues were obtained from patients who were diagnosed
with CRC and underwent surgery at Mianyang Central Hospital
(Mianyang, China) between January 2009 and December 2009. Patients
subjected to chemotherapy prior to surgery were excluded from the
current study. The tissues were obtained immediately after surgery
and frozen at −196°C in liquid nitrogen until further use. Written
informed consent was obtained from each patient at the day of
surgery, and the study was approved by the local Research Ethics
Committee of Mianyang Central Hospital. The clinicopathological
characteristics of patients are listed in Table I.
 | Table I.Clinicopathological variables in
patients with colorectal carcinoma. |
Table I.
Clinicopathological variables in
patients with colorectal carcinoma.
|
|
| Expression of
Sirt7 |
|
|---|
|
|
|
|
|
|---|
| Characteristics | No. | Low (n=21) | High (n=39) | P-value |
|---|
| Gender |
|
|
| 0.183 |
| Male | 27 | 7 | 20 |
|
|
Female | 33 | 14 | 19 |
|
| Age, years |
|
|
| 0.765 |
|
<50 | 27 | 10 | 17 |
|
| ≥50 | 33 | 11 | 22 |
|
| Tumor size
(diameter) |
|
|
| 0.009 |
| Small
(≤3 cm) | 29 | 15 | 14 |
|
| Large
(≥3 cm) | 31 | 6 | 25 |
|
| Tumor, node and
metastasis stage |
|
|
| 0.003 |
|
I–II | 30 | 16 | 14 |
|
|
III–IV | 30 | 5 | 25 |
|
| Lymph node
metastasis |
|
|
| 0.123 |
|
Absent | 29 | 13 | 16 |
|
|
Present | 31 | 8 | 23 |
|
| Distant
metastasis |
|
|
| 0.016 |
|
Absent | 22 | 12 | 10 |
|
|
Present | 38 | 9 | 29 |
|
| Tumor location |
|
|
| 0.417 |
|
Colon | 30 | 12 | 18 |
|
|
Rectum | 30 | 9 | 21 |
|
The tumor stage was defined using the seventh
edition of the tumor, node and metastasis (TNM) classification for
CRC of the American Joint Committee Cancer (14). Furthermore, lymph node metastasis was
identified using the lymph node pathological examination during the
surgery, and the distant metastasis was identified using abdominal
and pulmonary computed tomography prior to surgery. The diagnosis
of all patients was histopathologically confirmed. All the patients
were followed up to generate the overall survival times, with a
total follow-up period of 6 years (range, 1–6 years). The follow-up
information of all the participants was updated every 3 months by a
telephone conversation and questionnaire letters. The survival
times were calculated from the surgery date to the recurrence or
metastasis-associated mortality. Patients were divided into two
groups for assessing overall survival according to their median
Sirt7 mRNA expression levels, specifically the high and low Sirt7
expression groups. Sirt7 mRNA expression in frozen CRC tumor
tissues and paired adjacent non-tumor tissues was measured by
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR).
Cell culture
Human SW620 (CCL-227™) and SW480 (CCL-228™) colon
carcinoma cells were purchased from American Type Culture
Collection (ATCC; Manassas, VA, USA). In addition, HCT116 and HT29
colon cancer cell lines, as well as human normal colon epithelium
FHC cell line, were purchased from Shanghai Bioleaf Biotech Co.,
Ltd. (Shanghai, China). The aforementioned cells were cultured in
RPMI 1640 medium (Hyclone; GE Healthcare Life Sciences, Logan, UT,
USA) containing 10% fetal bovine serum (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) in a 5% CO2 atmosphere at 37°C.
Reagents and transfection
Sirt7 siRNAs were obtained from Sigma-Aldrich; Merck
KGaA. The sequence of the siRNA against Sirt7 (siRNA ID, SASI
Hs01_00133900) was 5′-CGCCAAATACTTGGTCGTCTA-3′ and CDH1
(E-cadherin; siRNA ID, SASI Hs01_00086310) was
5′-GAGATTGCACCGGTCGACAAAGCTC-3′, and the control siRNA sequence
[scramble control RNA (SCR)] was 5′-CCTAAGGTTAAGTCGCCCTCG-3′. A
final concentration of 50 nM Sirt7, 50 nM E-cadherin or 50 nM of
their corresponding negative control siRNA was used for transient
transfection with Lipofectamine 2000 (50 µl; Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) at a ratio of 1:1, and
the solutions were incubated for 20 min at room temperature.
Subsequently, the siRNA mixture was added to the cells and
incubated for 8 h at 37°C, according to the manufacturer's
protocol.
The Sirt7 full-length sequence was amplified from
human genomic DNA and cloned into the lentiviral vector GV208 using
the EcoRI and NotI sites (Genchem, Shanghai, China),
obtaining pGV-Sirt7. The primers for Sirt7 were as follows:
5′-ATATGAATTCGCCACCATGGCAGCCGGGGGTCTGATC-3′ (forward) and
5′-ATAAGGATGCGGCCGCTTACGTCACTTTCTTCCTTTTTGT-3′ (reverse). The
E-cadherin lentiviral expression vector was purchased from
Genchem.
293T cells (CRL-3216™; ATCC), used for virus
packaging, were cultured in Dulbecco's modified Eagle's medium in a
37°C incubator with 5% CO2. pGV-Sirt7, pHelper 1.0 and
pHelper 2.0 were cotransfected into 293T cells using
Lipofectamine® 2000. At 8 h after transfection, the
medium was refreshed, and at 48 h after transfection, the 25 ml
supernatant was harvested by centrifugation at 12,000 × g for 15
min at 4°C for further use. Empty lentiviral GV208 vector was used
as the negative control (pGV-NC). The SW620 and SW480 CRC cells
were infected with either pGV-Sirt7 or pGV-NC in the presence of 5
µg/ml Polybrene (Sigma-Aldrich; Merck KGaA). At 72 h
post-infection, the efficiency of infection was measured by
performing RT-qPCR. The E-cadherin lentiviral expression vector was
purchased from Genchem.
RNA isolation and RT-qPCR
Total RNA was isolated from the aforementioned cell
lines and tissues using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
purification and quantity of the RNA was measured with a NanoDrop
2000 spectrophotometer (Thermo Fisher Scientific, Inc.). Next, 1 µg
RNA was reverse-transcribed into cDNA using the TransGen First
Strand cDNA Synthesis kit (TransGen Biotech, Beijing, China). qPCR
was then performed using SYBR-Green Master Mix (Roche Applied
Science, Penzberg, Germany) on an Applied Biosystems Prism 7500
detection system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The amplification was performed according to the following
conditions: Denaturation at 95°C for 15 sec, and 40 cycles of
annealing at 60°C for 45 sec and extending at 72°C for 30 sec. The
primers used were as follows: E-cadherin forward,
5′-TGCTGCAGGTCTCCTCTTGG-3′ and reverse, 5′-AGTCCCAGGCGTAGACCAAG-3′;
Sirt7 forward, 5′-TACATTGAAGTCTGTACCTCC-3′ and reverse,
5′-GTGGGTACTTCTTTAGAACCT-3′; Vimentin forward
5′-ATTGAGATTGCCACCTACAG-3′ and reverse 5′-ATCCAGATTAGTTTCCCTCAG-3′;
GAPDH, forward 5′-GAGAAGTATGACAACAGCCTC-3′ and reverse
5′-ATGGACTGTGGTCATGAGTC-3′. The miRNA levels were normalized
against GAPDH and relative fold changes were calculated using the
2−ΔΔCq method (15).
Western blot analysis
Cells were lysed using radioimmunoprecipitation
assay buffer at 4°C for 45 min, and the protein concentration was
determined using a bicinchoninic acid assay kit (Thermo Fisher
Scientific, Inc.). Next, 30 µg protein was resolved with 10%
SDS-polyacrylamide gel electrophoresis and transferred to a
polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA). Following blocking with WB blocking solution (Beyotime
Institute of Biotechnology, Zhejiang, China), the membrane was
incubated overnight at 4°C with primary antibodies. The primary
antibodies used were from an epithelial-mesenchymal transition
(EMT) antibody kit (cat. no. 9782; Cell Signaling Technology, Inc.,
Danvers, MA, USA) in which each antibody was diluted at 1:1,000,
the SIRT7 antibody (cat. no. ab62748; 1:500; Abcam, Cambridge, MA,
USA) and the GAPDH antibody (cat. no. SC81545; 1:1,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) was used as an internal
control. Following three washes with extensive Tris-buffered
saline/Tween-20 (TBST) for 10 min, peroxidase-conjugated goat
anti-rabbit (sc-2004; 1:3,000) or goat anti-mouse IgG secondary
antibodies (sc-2005; 1:3,000) (both Santa Cruz Biotechnology, Inc.)
were used for incubation at room temperature for 1 h. Following the
washing of the membrane with TBST four times for 15 min, the
immunoreactivity was detected using an enhanced chemiluminescence
kit (Pierce; Thermo Fisher Scientific, Inc.) and quantified using
ImageJ software (version 2.1.4; National Institutes of Health,
Bethesda, MD, USA).
Cell proliferation assay
MTT and colony formation assays were performed to
measure the cell growth viability. For MTT assay, the transfected
cells were seeded into 96-well plate in triplicate, at a
concentration of 500 cells/well. At 24, 48, 72 and 96 h after
transfection, 20 µl MTT (5 mg/ml) was added to each well and
further incubated for 4 h at 37°C, followed by addition of 150 µl
of dimethyl sulfoxide to stop the reaction. The absorbance of each
well was measured at the wavelength of 570 nm on a microplate
reader in three independent experiments.
For the colony formation assay, the transfected
cells were seeded into 6-well plates in triplicate at a density of
1×103 cells/well and cultured for 10 days at 37°C.
Subsequent to fixing with 10% paraformaldehyde for 15 min at room
temperature, the colonies were stained with Giemsa for 30 min at
room temperature. Colonies with >50 cells were counted and
analyzed.
Cell invasion analysis
For the invasion assay, Transwell chambers precoated
with Matrigel were obtained from BD Biosciences (San Jose, CA,
USA). Transfected cells (2×104 cells per well) in
RPMI-1640 medium were added to the upper chambers. RPMI-1640 with
10% FBS was added to the lower chambers. At 24 h after
transfection, non-migrating cells on the upper side were gently
wiped off, while the cells that migrated through the filter were
fixed with 4% polyoxymethylene for 20 min at room temperature,
stained with 1% crystal violet for 30 min at room temperature
(Sigma-Aldrich; Merck KGaA) and counted using phase-contrast
microscopy.
Luciferase reporter assay
The luciferase reporter activity was conducted using
a Luciferase Assay system (Promega Corporation, Madison, WI, USA).
E-cadherin (−108)-Luc and Mutant E-cadherin (−108)-Luc were
generated as described previously (16). By transfecting the E-cadherin
reporter construct and Sirt7 or siSirt7 into the indicated cell
lines, and co-transfecting with pRL-SV40 renilla luciferase vector
as an internal control for transfection efficiency, luciferase
reporter activity was measured. Cells were harvested after 48 h and
lysates were assayed for luciferase activity, according to the
manufacturer's protocol. Luciferase activities were normalized to
renilla luciferase activity. Each experiment was performed in
triplicate.
Statistical analysis
All statistical analyses were performed using SPSS
version 17.0 software (SPSS, Inc., Chicago, IL, USA). Each
experiment was performed for at least three independent times and
the data were presented as the means ± standard deviation.
Differences were analyzed using χ2 test, Student's
t-test or one-way analysis of variance accordingly. Expression
comparisons between two groups were performed with a Student's
t-test. The χ2 test was used to evaluate the association
between Sirt7 expression and the clinicopathological
characteristics of patients. Cox log-rank test was used to test the
prognostic significance. P<0.05 was considered as an indicator
of a statistically significant difference.
Results
Expression of Sirt7 is upregulated in
CRC cell lines and tissues
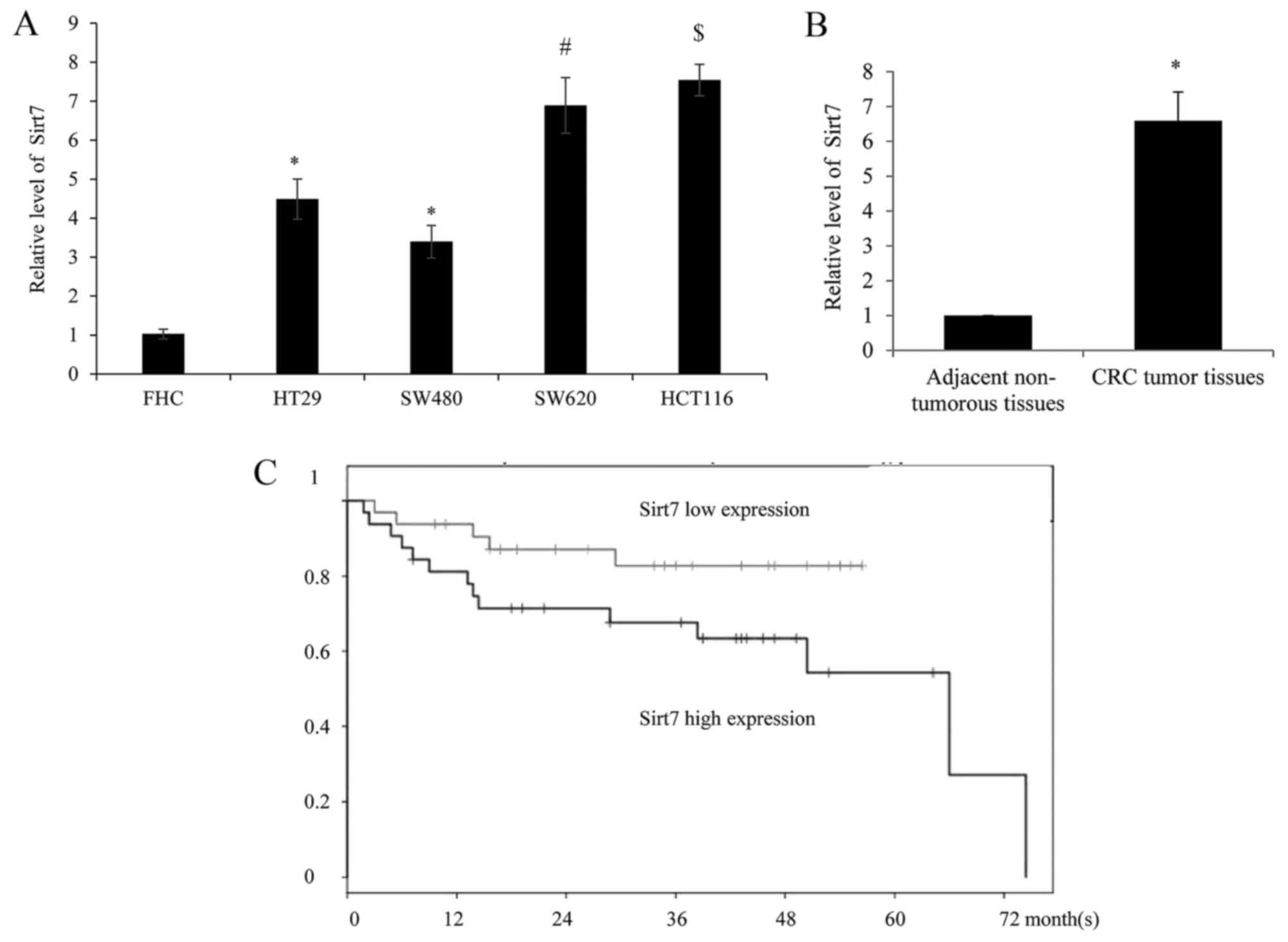
In order to examine the expression level of Sirt7 in
different CRC cell lines (HT29, SW480, SW620 and HCT116), along
with the human normal colorectal cell line FHC, the mRNA of cells
was harvested and analyzed by RT-qPCR. The results identified that
Sirt7 exhibited a significantly higher expression level in the CRC
cells as compared with the normal FHC cells (Fig. 1A). Furthermore, compared with the
low-metastatic tumor cells HT29 and SW480, a higher expression of
Sirt7 was detected in the highly-metastatic SW620 and HCT116 cells,
respectively.
RT-qPCR was also used to assess the expression of
Sirt7 in 60 CRC and adjacent non-tumorous tissues. As shown in
Fig. 1B, Sirt7 was significantly
upregulated in CRC tumor tissues compared with the corresponding
normal tissues. The clinical information of Sirt7 expression is
summarized in Table I, which
indicates that higher expression of Sirt7 was correlated with the
tumor size, TNM stage and distant metastasis in patients. However,
there was no statistically significant difference between the age,
gender, lymph node metastasis and tumor location, and the
expression of Sirt7. Furthermore, the association between Sirt7
expression and patient survival times was investigated. Depending
on the median Sirt7 expression level, the patients were divided
into the high (relative expression ≥2.57) and low (relative
expression <2.57) expression groups. Higher Sirt7 expression was
correlated with a worse overall survival rate, according to the
Kaplan-Meier curves, however all patients in the low expression
group succumbed prior to the 72 month follow-up (Fig. 1C).
Sirt7 exhibits oncogenic properties by
promoting CRC cell proliferation
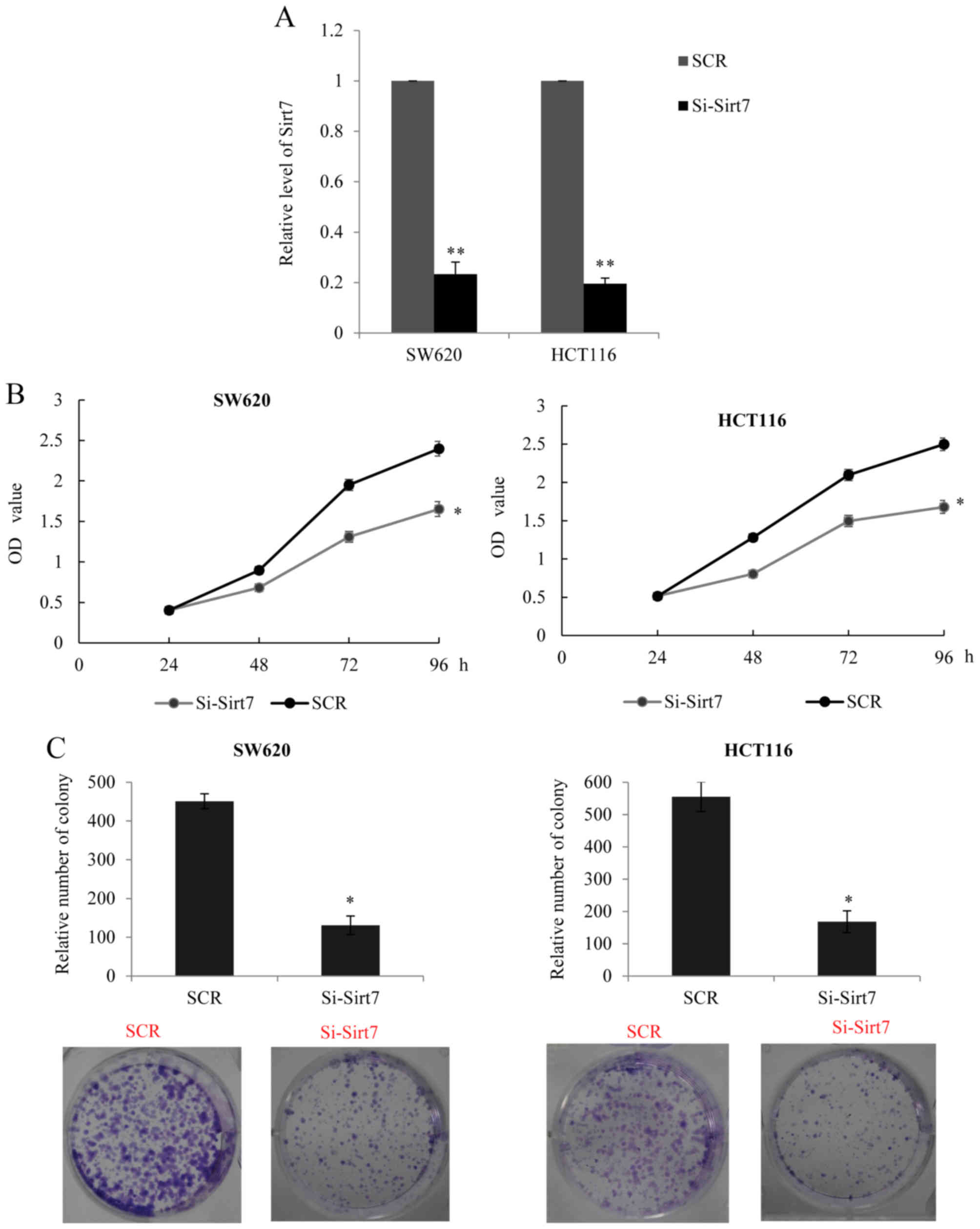
Since higher expression of Sirt7 was found to be
correlated with tumor size, it was hypothesized that Sirt7 may
promote CRC cell proliferation. In order to examine the role of
Sirt7, an RNA interference assay was performed to silence the
expression of Sirt7 (si-Sirt7 transfected group) in SW620 and
HCT116 cells. The transfection efficiency was analyzed using
RT-qPCR, and knockdown of Sirt7 was observed in the transfected
cells (Fig. 2A). Furthermore, the
MTT assay in the two cell lines indicated that silencing of Sirt7
may inhibit cancer cell proliferation (Fig. 2B). The colony formation assay also
revealed that the Sirt7 knockdown group had a lower colony-forming
efficiency in comparison with the control SCR group (Fig. 2C). The aforementioned data confirmed
that a decreased expression of Sirt7 may have a suppressive effect
on CRC cell proliferation.
Overexpression of Sirt7 increases the
invasion ability of CRC cells
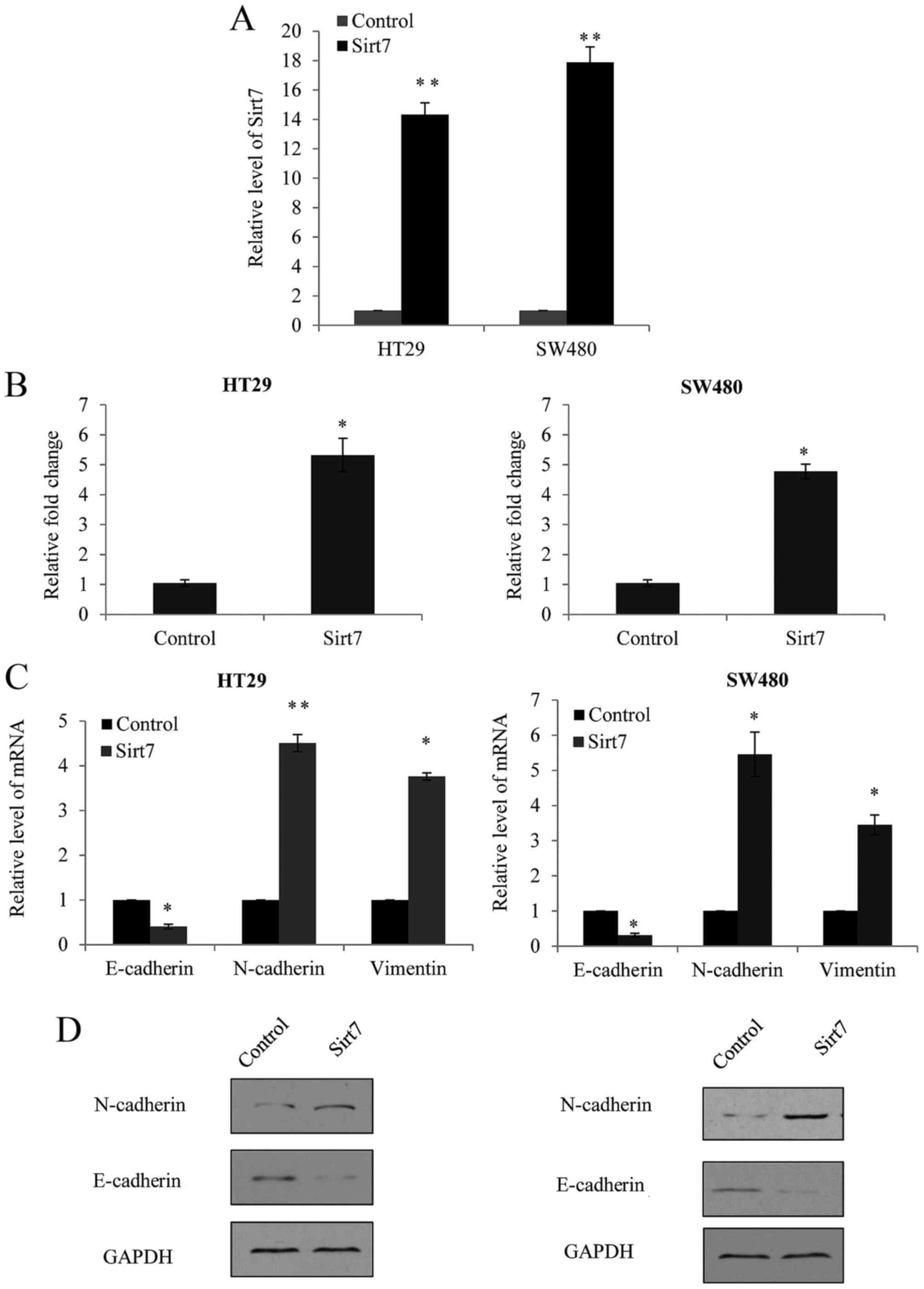
To investigate whether Sirt7 was involved in the CRC
invasion, a Sirt7-overexpression lentivirus was transfected into
HT29 and SW480 cells, which presented lower metastatic ability and
Sirt7 expression compared with the SW620 and HCT116 cells, as
observed earlier.
The efficiency of Sirt7 overexpression was assessed
by RT-qPCR. Empty lentiviral vector GV208, which did not contain
any external sequence, was used as the negative control (pGV-NC).
As shown in Fig. 3A, significant
overexpression of Sirt7 was observed in the lentivirus transfected
cells. Furthermore, a Transwell assay was performed in the HT29 and
SW480 cells, and cells transfected with the Sirt7 overexpression
vector exhibited a significantly increased ability of invasion in
the two cell lines (Fig. 3B).
As EMT is usually considered as the initial step of
metastasis, the present study detected the changes in the EMT
markers, E-cadherin, N-cadherin and vimentin. In HT29 and SW480
cells with overexpression of Sirt7, a marked upregulation of
N-cadherin and vimentin mRNA expression was observed, as well as
significant downregulation of E-cadherin (Fig. 3C). The changes in the expression
levels of these markers were also detected by western blot
analysis; however, since the antibody for vimentin is not
available, western blotting was not performed for vimentin. As
shown in Fig. 3D, increase of
N-cadherin and decrease of E-cadherin protein levels were observed
following Sirt7 overexpression. These findings supported the theory
that Sirt7 expression enhanced CRC EMT and invasion.
Sirt7 regulates E-cadherin
transcription in an E-box-dependent manner
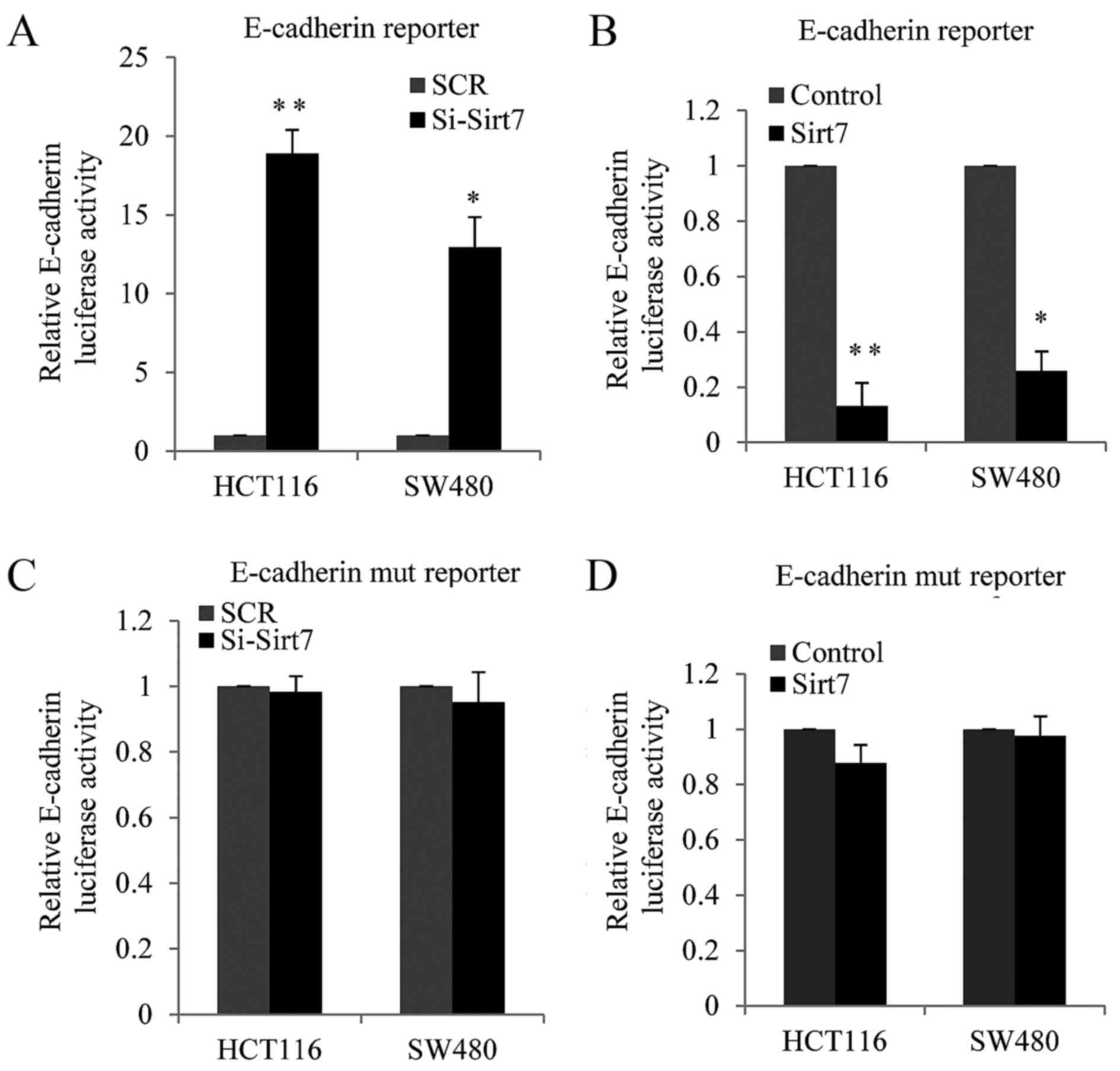
It is known that the overexpression of Sirt1
stimulates cell invasion by suppressing E-cadherin expression in
various cancer types (17,18). Thus, in the present study, it was
hypothesized that Sirt7, a new Sirt family member, may also
regulate E-cadherin. Luciferase assay was performed to examine the
function of Sirt7. An E-cadherin luciferase-reporter construct was
co-transfected along with si-Sirt7 (knockdown) or
Sirt7-overexpression vector and their corresponding controls into
HCT116 and SW480 cells. The results revealed that E-cadherin
luciferase activity was increased in the Sirt7-knockdown cells as
compared with the SCR cells (Fig.
4A). By contrast, when Sirt7 was overexpressed in the two cell
lines, the E-cadherin luciferase activity was decreased (Fig. 4B). Furthermore, E-box domain mutation
of E-cadherin was investigated in order to confirm whether the
inhibition of E-cadherin expression by Sirt7 was dependent on the
inhibition of the E-box.
Subsequently, co-transfection with si-Sirt7 and
E-box-mutated E-cadherin luciferase reporters was conducted in
HCT116 or SW480 cells, and the luciferase report activity was
measured. As shown in Fig. 4C, the
knockdown of Sirt7 had almost no effect on the E-box-mutated
E-cadherin luciferase reporter. HCT116 or SW480 cells were also
co-transfected with Sirt7-overexpression lentivirus and
E-box-mutated E-cadherin luciferase reporters in HCT116 cells or
SW480 cells. As shown from Fig. 4D,
Sirt7 exerted a reduced effect on the E-box-mutated E-cadherin
promoter compared with the E-box wild type promoter. These results
demonstrated that Sirt7 suppressed E-cadherin expression at the
transcriptional level in an E-box-dependent manner in the CRC cell
lines.
Sirt7 regulates CRC proliferation and
invasion in an E-cadherin-dependent manner
Based on the aforementioned results, the present
study further investigated whether Sirt7 regulates CRC
proliferation and metastasis in an E-cadherin-dependent manner. RNA
interference targeting the CDH1 gene, which encodes the E-cadherin
protein, was performed to silence the expression of E-cadherin in
SW620 and HCT116 cells. The transfection efficiency was analyzed
using RT-qPCR (Fig. 5A). In SW620
and HCT116 cells, E-cadherin was overexpressed using a lentivirus
and the efficiency of E-cadherin overexpression was assessed by
RT-qPCR, with an empty lentiviral GV208 vector used as a control
(Fig. 5B). The data demonstrated
that the siRNA and the E-cadherin overexpression lentiviruses were
successful and thus, were used for the analysis of Sirt7 function.
The MTT assay in the SW620 and HCT116 cells indicated that the
effect of Sirt7 knockdown on proliferation may be partially reduced
by the E-cadherin knockdown (Fig.
5C). A Transwell assay in the HT29 and SW480 cells also
demonstrated that, while E-cadherin was overexpressed in the cells
transfected with Sirt7-overexpression constructs, the invasion
ability was reduced (Fig. 5D). On
the basis of these results, it can be concluded that Sirt7
regulated CRC proliferation and invasion in an E-cadherin-dependent
manner.
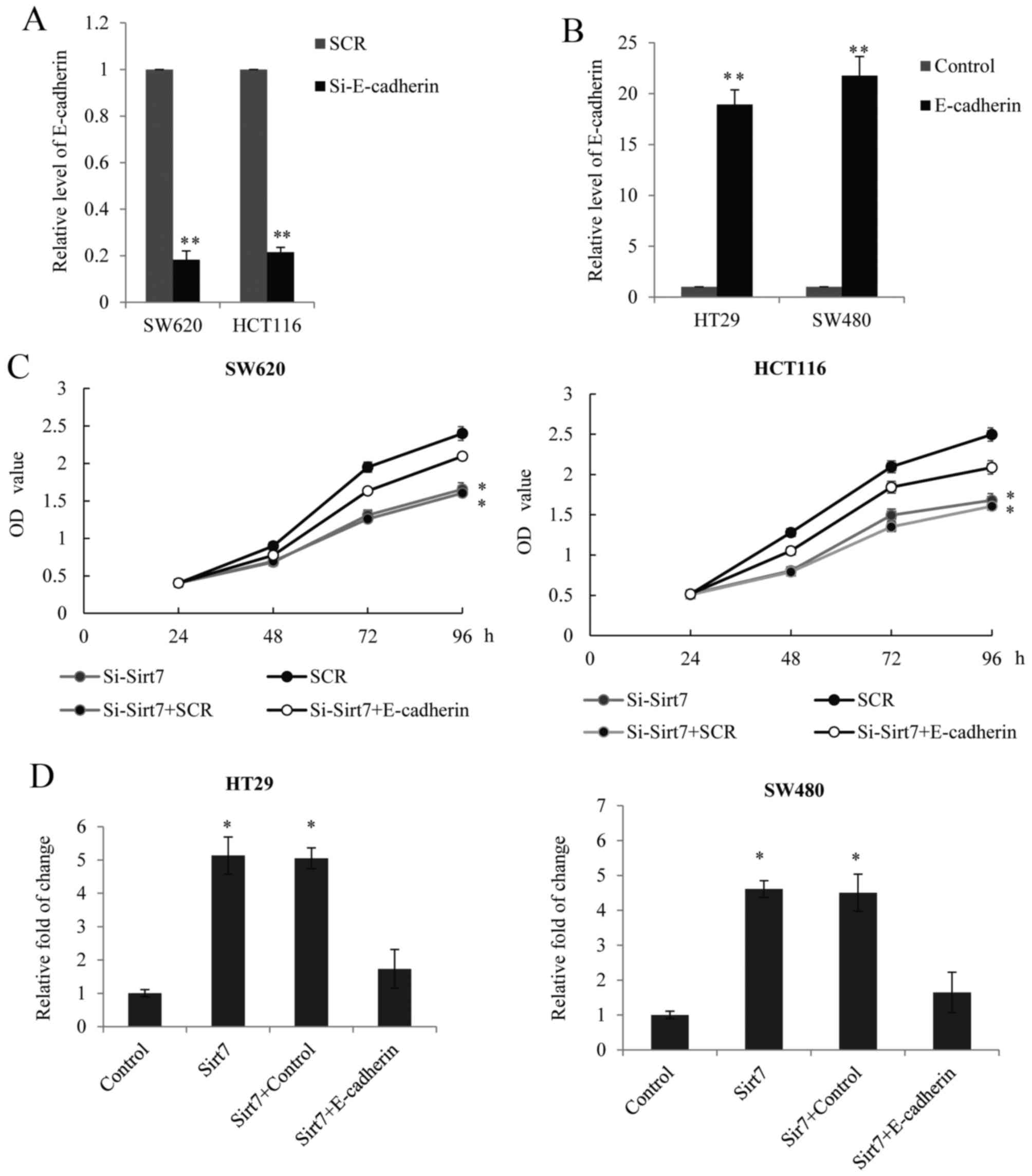 | Figure 5.Sirt7 regulated CRC proliferation and
invasion in an E-cadherin-dependent manner. Endogenous E-cadherin
level was measured in SW620 and HCT116 cells after transduction
with (A) E-cadherin siRNAs or SCR and (B) E-cadherin overexpression
lentivirus or lentiviral vector GV208 (control). GAPDH served as an
internal control. (C) MTT assay was performed in SW620 and HCT116
cells transfected with si-Sirt7, SCR, si-Sirt7+SCR or
si-Sirt7+si-E-cadherin. The OD value was measured every 24 h. (D)
Transwell assay was performed in HT29 and SW480 cells transfected
with Sirt7, vector, Sirt7+vector or Sirt7+E-cadherin. Cells
invading the lower chamber were stained and counted under a light
microscopy, and the results were presented as the fold change over
the vector. *P<0.05 and **P<0.01, vs. corresponding control
group. Sirt7, sirtuin 7; CRC, colorectal carcinoma; SCR, scramble
control RNA; si, siRNA. |
Discussion
Metastasis is one of the main hallmarks of cancer
and the leading cause of cancer-associated mortality (19). Epigenetic aberrations have been
demonstrated to contribute to the process of tumorigenesis and
metastasis in various ways (20).
Therefore, investigating the epigenetic regulation of CRC may
provide new insight into the underlying molecular mechanisms and
thus assist in the development of novel clinically relevant
prognostic biomarkers.
The Sirt family members target numerous key proteins
to modulate their state, from acetylation to deacetylation, and
have been reported to be involved in several pathological
conditions, including malignant tumors, cardiovascular disease and
diabetes (21–24). The function of Sirt1 in CRC has been
well discussed, and high Sirt1 expression has been reported to
enhance tumorigenesis and be associated with a poor prognosis of
CRC patients, including advanced-stage tumors and lymph node or
liver metastases (25,26). By targeting fos-related antigen 1,
Sirt1 promotes EMT and metastasis in CRC (27). While numerous studies on Sirt1 have
examined its biological properties, the expression panel and
function of Sirt7, a new member of the Sirt family, in human
malignancies remains unclear.
In recent years, emerging studies have been examined
the underlying mechanism of Sirt7 in promoting cancer development,
including hepatocellular carcinoma (28), bladder cancer (29) and ovarian cancer (4). Yu et al (30) reported that Sirt7 expression was
elevated in CRC tissues and cells, and was correlated with tumor
stage and poor prognosis. In Sirt7-overexpressing cells, vimentin
and fibronectin were upregulated, whereas E-cadherin and β-catenin
were downregulated (20), which was
consistent with the results of the present study; however, the
aforementioned study did not reveal the reasons underlying these
observations. In the present study, the initial aim was to uncover
the mechanism underlying the Sirt7 enhancing effect on CRC cell
proliferation and invasion, and shows evidence of the potential
rationale for Sirt7 as a therapeutic target. The current results
highlight that Sirt7 not only increased N-cadherin expression, but
also regulated E-cadherin transcription in an E-box-dependent
manner. Notably, although E-cadherin is traditionally considered as
a suppressor of cell invasion, the present study observed the
inhibitory function of E-cadherin on the cell proliferation,
similarly to the findings of Kim et al (31) and Ji et al (32) reported in CRC and pancreatic cancer.
Although the higher Sirt7 group had a worse overall survival rate,
the deaths of the lower Sirt7 group may be due to the complicated
tumor metastatic mechanism. Sirt7 is not the only oncogene for
CRC.
In conclusion, the present study demonstrated that
Sirt7 may serve as an oncogene and therapeutic target in patients
with CRC by directly inhibiting the expression of E-cadherin.
However, further studies are required to better define the
underlying molecular mechanisms and to explore the use of Sirt7
inhibitors in CRC. In addition, the function of Sirt7 in migration
should be further studied, as the migration activity was not
analyzed in the current study. Finally, a larger independent CRC
patient cohort is also necessary for the validation of these
results.
References
|
1
|
Kelly G: A review of the sirtuin system,
its clinical implications, and the potential role of dietary
activators like resveratrol: part 1. Altern Med Rev. 15:245–263.
2010.PubMed/NCBI
|
|
2
|
Kelly GS: A review of the sirtuin system,
its clinical implications, and the potential role of dietary
activators like resveratrol: part 2. Altern Med Rev. 15:313–328.
2010.PubMed/NCBI
|
|
3
|
Barber MF, Michishita-Kioi E, Xi Y,
Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL,
Chen K, et al: SIRT7 links H3K18 deacetylation to maintenance of
oncogenic transformation. Nature. 487:114–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang HL, Lu RQ, Xie SH, Zheng H, Wen XM,
Gao X and Guo L: SIRT7 Exhibits oncogenic potential in human
ovarian cancer cells. Asian Pac J Cancer Prev. 16:3573–3577. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang S, Chen P, Huang Z, Hu X, Chen M, Hu
S, Hu Y and Cai T: Sirt7 promotes gastric cancer growth and
inhibits apoptosis by epigenetically inhibiting miR-34a. Sci Rep.
5:97872015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geng Q, Peng H, Chen F, Luo R and Li R:
High expression of Sirt7 served as a predictor of adverse outcome
in breast cancer. Int J Clin Exp Pathol. 8:1938–1945.
2015.PubMed/NCBI
|
|
7
|
Singh S, Kumar PU, Thakur S, Kiran S, Sen
B, Sharma S, Rao VV, Poongothai AR and Ramakrishna G:
Expression/localization patterns of sirtuins (SIRT1, SIRT2, and
SIRT7) during progression of cervical cancer and effects of sirtuin
inhibitors on growth of cervical cancer cells. Tumour Biol.
36:6159–6171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ferrarotto R, Pathak P, Maru D, Agarwal A,
Overman M, Hoff PM and Kopetz S: Durable complete responses in
metastatic colorectal cancer treated with chemotherapy alone. Clin
Colorectal Cancer. 10:178–182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kraus S, Nabiochtchikov I, Shapira S and
Arber N: Recent advances in personalized colorectal cancer
research. Cancer Lett. 347:15–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park IJ and Yu CS: Current issues in
locally advanced colorectal cancer treated by preoperative
chemoradiotherapy. World J Gastroenterol. 20:2023–2029. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Andre N and Schmiegel W: Chemoradiotherapy
for colorectal cancer. Gut. 54:1194–1202. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O'Connell JB, Maggard MA and Ko CY: Colon
cancer survival rates with the new American Joint Committee on
Cancer sixth edition staging. J Natl Cancer Inst. 96:1420–1425.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hajra KM, Chen DY and Fearon ER: The SLUG
zinc-finger protein represses E-cadherin in breast cancer. Cancer
Res. 62:1613–1618. 2002.PubMed/NCBI
|
|
17
|
Deng S, Zhu S, Wang B, Li X, Liu Y, Qin Q,
Gong Q, Niu Y, Xiang C, Chen J, et al: Chronic pancreatitis and
pancreatic cancer demonstrate active epithelial-mesenchymal
transition profile, regulated by miR-217-SIRT1 pathway. Cancer
Lett. 355:184–191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Byles V, Zhu L, Lovaas JD, Chmilewski LK,
Wang J, Faller DV and Dai Y: SIRT1 induces EMT by cooperating with
EMT transcription factors and enhances prostate cancer cell
migration and metastasis. Oncogene. 31:4619–4629. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ellis L, Atadja PW and Johnstone RW:
Epigenetics in cancer: Targeting chromatin modifications. Mol
Cancer Ther. 8:1409–1420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ashraf N, Zino S, Macintyre A, Kingsmore
D, Payne AP, George WD and Shiels PG: Altered sirtuin expression is
associated with node-positive breast cancer. Br J Cancer.
95:1056–1061. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang L, Ren X, Cheng Y, Huber-Keener K,
Liu X, Zhang Y, Yuan YS, Yang JW, Liu CG and Yang JM:
Identification of Sirtuin 3, a mitochondrial protein deacetylase,
as a new contributor to tamoxifen resistance in breast cancer
cells. Biochem Pharmacol. 86:726–733. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dong XC: Sirtuin biology and relevance to
diabetes treatment. Diabetes Manag (Lond). 2:243–257. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Calabrese V, Cornelius C, Leso V,
Trovato-Salinaro A, Ventimiglia B, Cavallaro M, Scuto M, Rizza S,
Zanoli L, Neri S and Castellino P: Oxidative stress, glutathione
status, sirtuin and cellular stress response in type 2 diabetes.
Biochim Biophys Acta. 1822:729–736. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen X, Sun K, Jiao S, Cai N, Zhao X, Zou
H, Xie Y, Wang Z, Zhong M and Wei L: High levels of SIRT1
expression enhance tumorigenesis and associate with a poor
prognosis of colorectal carcinoma patients. Sci Rep. 4:74812014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lv L, Shen Z, Zhang J, Zhang H, Dong J,
Yan Y, Liu F, Jiang K, Ye Y and Wang S: Clinicopathological
significance of SIRT1 expression in colorectal adenocarcinoma. Med
Oncol. 31:9652014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheng F, Su L, Yao C, Liu L, Shen J, Liu
C, Chen X, Luo Y, Jiang L, Shan J, et al: SIRT1 promotes
epithelial-mesenchymal transition and metastasis in colorectal
cancer by regulating Fra-1 expression. Cancer Lett. 375:274–283.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim JK, Noh JH, Jung KH, Eun JW, Bae HJ,
Kim MG, Chang YG, Shen Q, Park WS, Lee JY, et al: Sirtuin7
oncogenic potential in human hepatocellular carcinoma and its
regulation by the tumor suppressors MiR-125a-5p and MiR-125b.
Hepatology. 57:1055–1067. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Han Y, Liu Y, Zhang H, Wang T, Diao R,
Jiang Z, Gui Y and Cai Z: Hsa-miR-125b suppresses bladder cancer
development by down-regulating oncogene SIRT7 and oncogenic long
non-coding RNA MALAT1. FEBS Lett. 587:3875–3882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu H, Ye W, Wu J, Meng X, Liu RY, Ying X,
Zhou Y, Wang H, Pan C and Huang W: Overexpression of sirt7 exhibits
oncogenic property and serves as a prognostic factor in colorectal
cancer. Clin Cancer Res. 20:3434–3445. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim S, Ahn SH, Yang HY, Lee JS, Choi HG,
Park YK and Lee TH: Modification of cysteine 457 in plakoglobin
modulates the proliferation and migration of colorectal cancer
cells by altering binding to E-cadherin/catenins. Redox Rep.
22:272–281. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ji M, Fan D, Yuan L, Zhang Y, Dong W and
Peng X: EBP50 inhibits pancreatic cancer cell growth and invasion
by targeting the beta-catenin/E-cadherin pathway. Exp Ther Med.
10:1311–1316. 2015. View Article : Google Scholar : PubMed/NCBI
|