Introduction
Liver injury or dysfunction is recognized as a
serious worldwide health problem. Clinically available synthetic
drugs for the treatment of liver diseases, such as interferon and
corticosteroids, are expensive, particularly for patients in
developing countries. These drugs may also cause adverse reactions
and further damage (1). Therefore,
traditional medicine is important in the treatment of liver
diseases (2). Although traditional
medicinal treatments have achieved good results, numerous problems
remain due to the complexity of a single herbal component, the
diversity of combination drugs, the uncertainty of drug
formulation, randomness of oral administration and unknown
mechanisms of action. Effective drugs with a clear mechanism and
low incidence of side effects are urgently required.
Carbon tetrachloride (CCl4) has been
widely used to induce chronic and acute liver damage in animal
models (3). Liver damage caused by
CCl4 is characterized by inflammation, formation of
trichloromethyl radicals and overproduction of reactive oxygen
species (ROS), which initiate lipid peroxidation and finally lead
to hepatotoxicity (4).
Anti-apoptotic, anti-oxidant and anti-inflammatory actions may be
important in the protection against CCl4-induced liver
damage.
Oxidative damage caused by ROS may lead to various
human diseases, such as liver fibrosis, cancer and inflammation. It
is well known that oxidative stress is involved in the pathogenesis
of acute and chronic liver injury (5). Hepatic damage caused by viral
infection, ethanol ingestion, iron overload and exposure to drugs
or CCl4 is attributed to overproduction of ROS (6,7). NADPH
oxidases (NOXs), which have a critical role in the inflammatory
response, contribute to ROS production during liver injury
(8).
The mitogen-activated protein kinase (MAPK) family
is involved in the regulation of cell proliferation and death in
response to various internal stresses. P38 MAPK and c-Jun
N-terminal kinase (JNK), two members of the MAPK superfamily, are
activated by cytokines such as tumor necrosis factor (TNF)-α and
interleukin (IL)-1β, or G protein-coupled receptors, and have an
important role in inflammation and apoptosis in response to stress
(9). JNKs have a vital role in the
death receptor-initiated extrinsic and mitochondrial intrinsic
apoptotic pathways (10). JNKs
activate apoptotic signaling by upregulating pro-apoptotic genes
via the transactivation of specific transcription factors or by
modulating the activities of mitochondrial pro- and anti-apoptotic
proteins through distinct phosphorylation (11). ROS may cause apoptosis by activating
the JNK signaling pathway (12).
CCl4 has been found to induce hepatic apoptosis via the
mitochondrial intrinsic and extrinsic apoptotic pathways (13,14). p38
MAPK has an essential role in regulating numerous cellular
processes, including inflammation and apoptosis. In turn,
production of p38 MAPK may be induced by inflammatory factors and
stress. CCl4 significantly increases the levels of p38
MAPK, and oxidative stress as well as certain cytokines activate
p38 MAPK through phosphorylation (15). The activation of the p38 MAPK pathway
accelerates cell apoptosis (16).
Studies have confirmed that certain factors that activate JNKs also
activate p38 MAPK (16,17). Furthermore, CCl4 was
reported to induce apoptosis in the liver by modulating the JNK and
p38 MAPK pathways. JNK regulates the expression of pro- and
anti-apoptotic members of the Bcl-2 family such as B-cell lymphoma
2 (Bcl-2) and Bcl-2-associated X protein (Bax). p38 MAPK induces
Bax translocation and enhances the expression of TNF-α to
ultimately induce apoptosis (10,18). In
response to extrinsic as well as intrinsic apoptotic stimuli, JNK
and P38 MAPK have an important role by interacting and modulating
the activities of caspase proteins (12,19).
Caspase-3 is one of the critical executioners of apoptosis, capable
of cleaving or degrading numerous key proteins such as nuclear
lamins, fodrin and the nuclear enzyme poly (adenosine diphosphase
ribose) polymerase (PARP) (12,20).
Methyl ferulic acid (MFA) is a monomer that is
extracted and purified from Securidaca inappendiculata
Hasskarl (21–23), which was traditionally used for the
treatment of acute or chronic hepatitis and exhibited some
inhibitory effects on hepatitis B surface antigen in T cell lines
(24). However, only few studies
have assessed the hepatoprotective effect of MFA (24). The present study investigated the
effects of MFA on CCl4-induced acute liver injury in
rats. Specifically, the inhibitory effect of MFA on inflammation,
oxidative stress and apoptosis was assessed, as well as the
involvement of p38 MAPK and JNK signaling.
Materials and methods
Animals
A total of 60 Sprague Dawley (SD) rats ((8–10 weeks;
30 males and 30 females) weighing 250–300 g were obtained from the
Experimental Animal Center of Guilin Medical University (Guilin,
China). The rats were kept in an environmentally controlled room
with a temperature of 25±2°C, relative humidity of 55±10% and a
12-h light/dark cycle. The rats were allowed free access to food
and water. The SD rats were randomly divided into six groups (n=10
in each). Rats in the control group and the CCl4-treated
model group only received an equivalent of distilled water
containing 0.1% Tween 80 by oral gavage once a day for one week.
Rats in the dimethyl diphenyl bicarboxylate (DDB)-treated group
(positive control group) received DDB in distilled water containing
0.1% Tween 80 at a dose of 200 mg/kg body weight by oral gavage
once a day for one week. Low, medium and high MFA-treated groups
received MFA in distilled water containing 0.1% Tween 80 at a dose
of 25, 50 or 100 mg/kg body weight by oral gavage once a day for a
week. One hour after the last treatment, all rats in the
CCl4-treated model group, the DDB-treated group and the
MFA-treated group received an intraperitoneal injection of
CCl4 (1 ml/kg body weight), while the control group
received an equivalent volume of 0.9% physiological saline solution
instead. At 24 h after CCl4 treatment, all rats were
sacrificed and a portion of liver tissues was immediately collected
for analysis and placed in ice-cold 0.9% physiological saline
solution to remove blood cells for ROS detection. The remaining
liver tissues were immediately stored at −80°C for later use. The
present study was performed in accordance with the Chinese
legislation and the US National Institutes of Health guidelines for
the use and care of experimental animals. All animal experiments
were approved by the institutional ethical committee of Guilin
Medical University (Guilin, China).
Measurement of serum aminotransferase
activities
After blood collection, serum was separated by
centrifugation at 3,200 × g for 20 min at room temperature. The
activities of alanine aminotransferase (ALT) and aspartate
aminotransferase (AST) in serum from rats were determined using
commercially available diagnostic kits (Alanine aminotransferase
assay kit; cat no. C009-2; Aspartate aminotransferase assay kit;
cat no. C010-2; Nanjing Jiancheng Bio Co., Ltd., Nanjing, China)
according to the manufacturer's instructions.
Assay of hepatic levels of superoxide
dismutase (SOD), glutathione peroxidase (GSH-Px), malondialdehyde
(MDA) and catalase (CAT)
Liver tissue samples were homogenized in nine
volumes of ice-cold 50 mM phosphate buffer (pH 7.4) and centrifuged
at 3,200 × g for 20 min at 4°C. Supernatants were used to determine
SOD, GSH-Px, MDA, CAT and total protein concentrations by using
commercially available diagnostic kits (SOD assay kit; cat no.
A001-3; GSH-PX assay kit; cat no. A005; cat no. MDA assay kit; cat
no. A003-1; CAT assay kit; cat no. A007-1; total protein assay kit;
cat no. A045-3; Nanjing Jiancheng Bio Co., Ltd.). The levels of
MDA, GSH-Px, SOD and CAT were normalized to the content of total
protein.
Hematoxylin and eosin (H&E)
staining
For histological examination, liver tissues were
removed from a portion of the left lobe and fixed in 10%
phosphate-buffered formalin. After being processed by routine
histological procedures, the samples were cut into 5-µm slices.
Sections were stained using hematoxylin for 5 min at 40°C and eosin
solution for 1 min at room temperature, then the slides were
observed for conventional morphological evaluation under a light
microscope (BX41; Olympus, Tokyo, Japan) and images were captured
at ×100 magnification. The degree of hepatic damage was evaluated.
Histological changes were scored according to the following system:
0, no injury; 1, mild injury; 2, moderate injury; and 3, severe
injury.
Semi-quantitative polymerase chain
reaction (qPCR)
Total RNA was extracted from liver tissues using a
tissue total RNA isolation kit (cat no. B518651; Shanghai Sangong
Pharmaceutical Co., Ltd., Shanghai, China) according to the
manufacturer's protocol. Total RNA was reversibly transcribed into
complementary DNA (cDNA) using a cDNA synthesis kit (TIANScript
MMLV; cat no. ER104; Tiangen Biotech Co., Ltd., Beijing, China)
according to the manufacturer's protocol. An MJ PTC-200 PCR System
(Bio-Rad, Hercules, CA, USA) and a qPCR kit (cat no. PC0902; 2× Taq
PCR Master Mix; Aidlab Biotechnologies Co., Ltd., Beijing, China)
were used based on the manufacturer's instructions for
amplification of target genes. The primers used in the study are
listed in Table I. The specific
primers for target gene β-actin were synthesized by Sangon Biotech
Co., Ltd. (Shanghai, China). As an internal standard control, the
expression level of β-actin was simultaneously quantified. The PCR
protocol was as follows: Initial denaturation for 3 min at 94°C;
30–40 cycles of denaturation for 30 sec at 94°C, annealing for 30
sec at 56–58°C, and extension for 1 min at 70°C; and a final
extension for 5 min at 72°C. The PCR products were identified by
electrophoresis using 1.5% agarose gel, and optical density of
target gene bands was calculated in each sample using a Gel Doc XR+
automatic gel imaging analysis system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and with adjustment through β-actin correction
to finally obtain the relative expression of target gene in each
sample (25).
 | Table I.Primer sequences used for the
determination of NOX4, p22phox, TNF-α, IL-1β and β-actin
gene expression. |
Table I.
Primer sequences used for the
determination of NOX4, p22phox, TNF-α, IL-1β and β-actin
gene expression.
| Genes | Oligonucleotide
primer sequences (5′-3′) | Product length
(bp) |
|---|
| NOX4 | Forward,
TGTGCCGAACACTCTTGGC | 136 |
|
| Reverse,
ATATGCACGCCTGAGAAAATA |
|
|
p22phox | Forward,
TATTGTTGCAGGAGTGCTCA | 103 |
|
| Reverse,
CACAGCGGTCAGGTACTTCT |
|
| TNF-α | Forward,
GGCAGGTCTACTTTGGAGTC |
|
|
| Reverse,
GCAGGCAGTATCACTCATTG | 233 |
| IL-1β | Forward,
GCAGGCAGTATCACTCATTG |
|
|
| Reverse,
CACACCAGCAGGTTATCATC | 165 |
| β-actin | Forward,
GACTCCTATGTGGGTGACGA | 199 |
|
| Reverse,
ACGGTTGGCCTTAGGGTTCA |
|
Western blot analysis
Total protein was extracted from liver tissues with
radio immunoprecipitation assay lysis buffer (cat no. P0013B,
Beyotime Institutute of Biotechnology, Shanghai, China). The
Mitochondrial protein was extracted from liver tissue using a
Cytoplasmic and Mitochondrial Protein Extraction kit (cat no.
C500051; Sangon Biotech Co., Ltd., Shanghai, China). Protein
concentration was determined using a bicinchoninic acid assay kit
(Beyotime Biotechnology, Inc.). A total of 50 µg/lane of sample
proteins were separated by 12% SDS-PAGE (Bio-Rad Laboratories, Inc.
USA). The separated proteins were then transferred to pure
nitrocellulose blotting membranes. The membranes were then blocked
for 1 h with 5% bovine serum albumin (cat no. B600036; Sangon
Biotech Co., Ltd., Shanghai, China) in Tris-buffered saline
containing 0.05% Tween 20 (TBST) at room temperature. The membranes
were incubated with anti-NOX4 (1:500; cat no. D121050), anti-Bcl-2
(1:1,000; cat no. D151442), anti-caspase-3 (1:1,000; cat no.
D220074), anti-cleaved caspase-3 (1:500; cat no. D260009), anti-Bax
(1:1,000; cat no. D220073), anti-GAPDH (1:1,000; cat no. D110016;
all from Sangon Biotech Co., Ltd., Shanghai, China),
anti-p22phox (1:500; cat no. BS60290; Bioworld
Technology, Inc., Nanjing, China), anti-phospho-p38 MAPK (1:1,000;
cat no. 4511T), anti-p38 MAPK (1:1,000; cat no. 14451),
anti-phospho-JNK (1:1,000; cat no. 4668T), anti-JNK (1:1,000; cat
no. 9252T; all from Cell Signaling Technology, Inc., Danvers, MA,
USA) or anti-IL-1β (1:1,000; cat no. sc-52012), anti-TNF-α
(1:1,000; cat no. sc-33639; both from Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) primary antibodies at 4°C overnight. The
samples were incubated with corresponding horseradish
peroxidase-conjugated secondary antibodies (1:50,000; cat no.
ZB2301; horseradish peroxidase (HRP) Affinipure Goat anti-rabbit
immunoglobulin G or 1:50,000; ZB2305 HRP Affinipure Goat anti-Mouse
immunoglobulin G; Zhongshang Goldenbridge Bio, Beijing, China) at
room temperature for one hour and protein bands were visualized by
enhanced chemiluminescence (cat no. E002-100; 7seapharmatech Co.
Ltd, Shanghai, China). The imaging system Chemi Doc XRS+ (Bio-Rad
Laboratories, Inc., USA) was used for imaging and quantitative
analysis of the blots. VCDA1 or GAPDH protein was used as an
internal control.
Fluorescent spectrophotometry
The level of ROS was determined by detecting the
fluorescence intensity of the oxidant-sensitive probe
2,7-dichlorodihydrofluorescein diacetate (Molecular Probes; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) as described in a
previous study (26). The amount of
formed dichlorofluorescein in the clear supernatant was determined
using a microplate reader (Infinite M200 PRO; Tecan, Zurich,
Switzerland) at an excitation wavelength of 502 nm and an emission
wavelength of 523 nm.
Thiobarbituric acid (TBA) reactive
substances (TBARS) colorimetric assay
Tissue lipid peroxidation was measured using a TBARS
colorimetric assay. Liver homogenate was incubated with 8.1% (w/v)
SDS for 10 min, followed by addition of 20% acetic acid (pH 3.5).
The reaction mixture was incubated with 0.6% TBA (w/v) for 1 h in a
boiling water bath. Pink color chromogen was extracted in
butanol-pyridine solution (15:1) and spectrophotometrically
quantified at 532 nm.
Measurement of the total anti-oxidant
capacity (TAC)
Based on the oxidation of intracellular
anti-oxidants with iron (III) in acidic medium, the TAC in the
liver was assayed with a commercially available assay kit (cat no.
A015-1; Nanjing Jiancheng Bio Co., Nanjing, China). The TAC of the
samples was measured according to the manufacturer's protocol. One
unit of TAC was defined as the capability of increasing the optical
density value at 520 nm by 0.01 per mg protein per min at 37°C.
Statistical analysis
All statistical analyses were performed using SPSS
software (version 17.0; International Business Machines, Corp.,
Armonk, NY, USA). One-way analysis of variance was used to
determine significant differences between groups. The
Student-Newman-Keuls test was used for comparisons between groups.
Values are expressed as the mean ± standard deviation. P<0.05
was considered to indicate a statistically significant
difference.
Results
MFA provides protection against
CCl4-induced hepatic injury
To determine whether MFA attenuates liver damage in
CCl4-treated rats, the activities of ALT and AST in
serum were measured. Compared with those in the normal control, the
activities of ALT and AST in serum from the model group were
significantly increased (P<0.01). Of note, administration of MFA
at all doses significantly inhibited the elevation of ALT levels,
and MFA at 50 and 100 mg/kg significantly inhibited the elevation
of AST levels in CCl4-treated rats in a dose-dependent
manner (P<0.05; Table II). These
results suggested that MFA provides protection against
CCl4-induced liver injury.
| Table II.Effect of MFA administration on ALT
and AST activities in serum of rats with liver damage induced by
CCl4. |
Table II.
Effect of MFA administration on ALT
and AST activities in serum of rats with liver damage induced by
CCl4.
| Group | ALT (U/l) | AST (U/l) |
|---|
| Control |
23.85±9.50 |
61.14±17.35 |
| Model |
216.39±70.93a |
524.01±160.71a |
| DDB (200
mg/kg) |
130.69±41.33b |
360.28±102.76b |
| MFA (25 mg/kg) |
170.56±51.56c |
465.18±137.63 |
| MFA (50 mg/kg) |
150.72±36.99b |
380.04±111.66b |
| MFA (100
mg/kg) |
134.72±37.52b |
353.54±109.15b |
MFA suppresses CCl4-induced
oxidative liver injury
To quantify oxidative liver injury, the hepatic
levels of SOD, GSH-Px, MDA and CAT were assayed. The hepatic levels
of MDA were assessed as an indicator of lipid peroxidation in
oxidative liver damage, and CCl4 treatment obviously
increased the hepatic MDA levels compared with those in the control
group (P<0.01), which was significantly inhibited by
pre-administration of MFA (P<0.05; Table III). Furthermore, the results
demonstrated that the activities of SOD, GSH-Px and CAT in the
model group were significantly decreased compared with those in the
control group, but pre-treatment with DDB or MFA (50 or 100 mg/kg)
significantly increased the activities of SOD, GSH-Px and CAT
compared with those in the model group (P<0.01). In addition,
CCl4 treatment significantly increased hepatic MDA
levels compared with those in the control group (P<0.01), but
pre-treatment with DDB or MFA significantly decreased MDA levels
compared with those in the model group (P<0.05). Of note, the
low dose of MFA had no significant effect on SOD or GSH-Px
(Table III). These results
indicated that MFA suppressed CCl4-induced oxidative
liver injury.
| Table III.Effect of MFA administration on SOD,
CAT and GSH-Px activities as well as the level of MDA in liver
tissues of rats induced by CCl4. |
Table III.
Effect of MFA administration on SOD,
CAT and GSH-Px activities as well as the level of MDA in liver
tissues of rats induced by CCl4.
| Group | SOD (U/mg
prot) | CAT (U/mg
prot) | GSH-Px (U/mg
prot) | MDA (nmol/g
prot) |
|---|
| Control |
5.14±1.36 |
66.70±6.16 |
515.36±133.47 |
22.78±7.63 |
| Model |
2.17±0.74a |
36.31±7.29a |
307.05±85.33a |
45.78±11.92a |
| DDB (200
mg/kg) |
4.30±0.79b |
59.79±6.21b |
492.07±127.63b |
32.55±9.50b |
| MFA (25 mg/kg) |
2.81±1.00 |
50.33±6.29b |
396.12±109.76 |
36.53±9.59c |
| MFA (50 mg/kg) |
3.34±0.72b |
60.37±5.49b |
456.87±131.95b |
32.43±6.52b |
| MFA (100
mg/kg) |
4.38±0.95b |
62.01±5.44b |
495.18±116.97b |
26.78±4.94b |
MFA alleviates CCl4-induced
histological changes in the liver
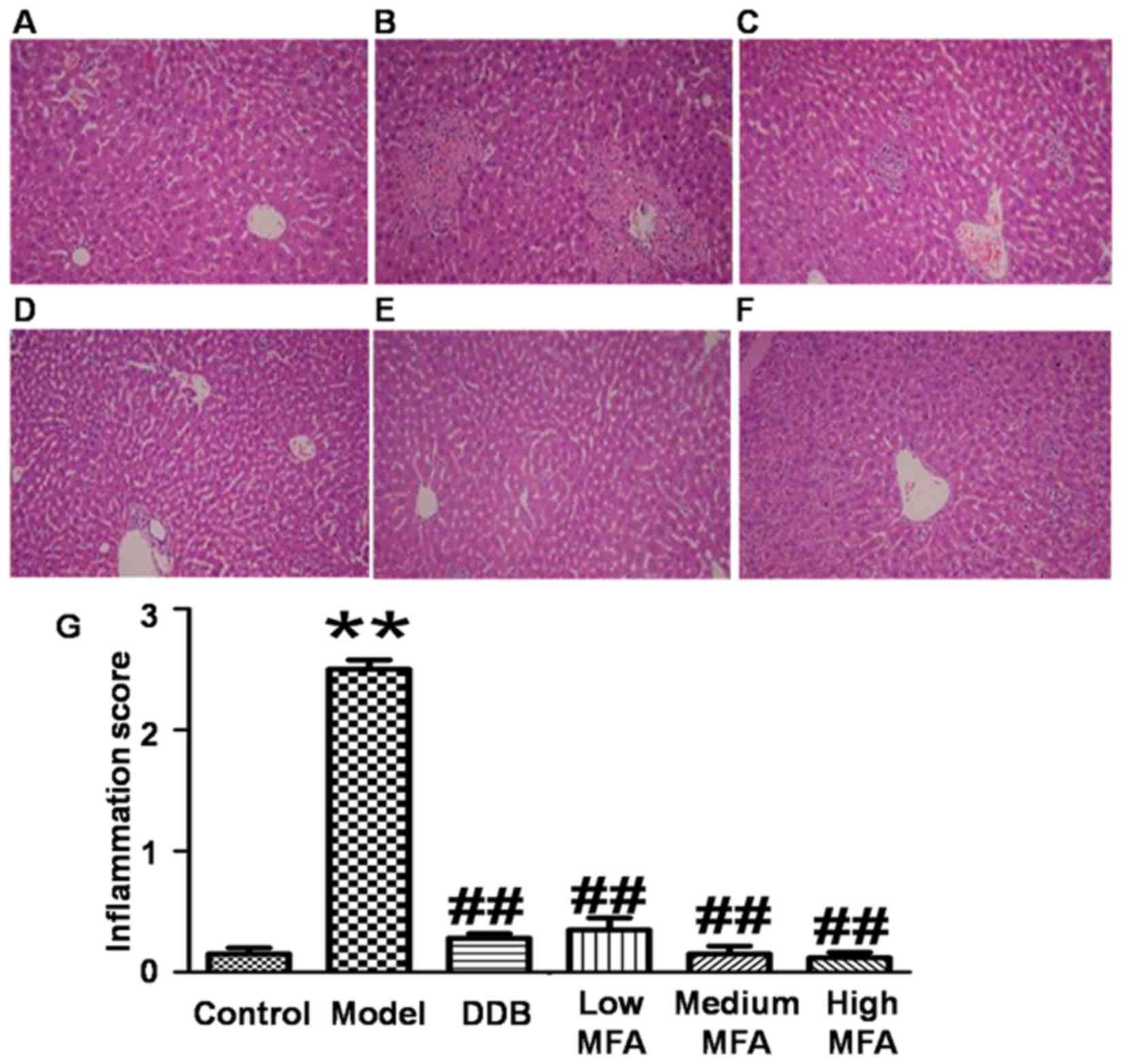
To detect histological changes in the liver, H&E
staining was performed. Visual observation revealed that livers
from the normal control group were reddish brown, soft and elastic,
but livers from the CCl4 model group exhibited
significantly increased liver volume, blood stasis, edge thickening
and liver surface with petechial hemorrhage. In the MFA (25 mg/kg)
group, the liver was slightly enlarged, and spot bleeding was
significantly reduced. By contrast, the appearance of the liver in
the medium and high MFA groups was close to normal. H&E
staining of liver sections from the normal control group
demonstrated an intact hepatic lobular structure, normal hepatic
cells with well-preserved cytoplasm, prominent nuclei, hepatocytes
that were radially arranged around the central vein, well-defined
sinusoidal line, uniform size, no degeneration, no necrosis, and
hepatic cords that were arranged in neat rows. In addition, no
inflammatory cell infiltration was observed (Fig. 1A). In the model group treated with
CCl4, typical pathological characteristics were
observed, including destroyed hepatic lobule structure, liver sinus
and central venous dilatation, hyperemia, a disorder in the
arranged of hepatic cords, ballooning degeneration, broad
infiltration of inflammatory cells, centrilobular fatty changes,
apoptosis and widespread hepatocellular necrosis, particularly
significant bridging necrosis, and inflammatory cell infiltration
in hepatic lobules and portal area (Fig.
1B). By contrast, CCl4-intoxicated rats pre-treated
with DDB had nearly normal liver tissues with no significant
changes in hepatocytes (Fig. 1C). In
the low MFA group, liver sections exhibited moderate hypertrophy of
hepatocytes with a relatively intact central vein, spotty necrosis,
a rare large area of necrosis, shrinking sinusoidal line and
reduced number of inflammatory cells (Fig. 1D). Of note, hepatic lesions were
markedly ameliorated in the medium and high MFA groups, with slight
inflammatory cell infiltration (Fig. 1E
and F). The inflammation score of CCl4-treated rats
was significantly higher than that of normal control rats, while
pre-treatment with MFA reduced the inflammation score (Fig. 1G). These results indicated that
CCl4 treatment caused obvious histological changes in
the liver, while pre-treatment with MFA prevented
CCl4-induced damage.
MFA inhibits CCl4-induced
oxidative stress in the liver
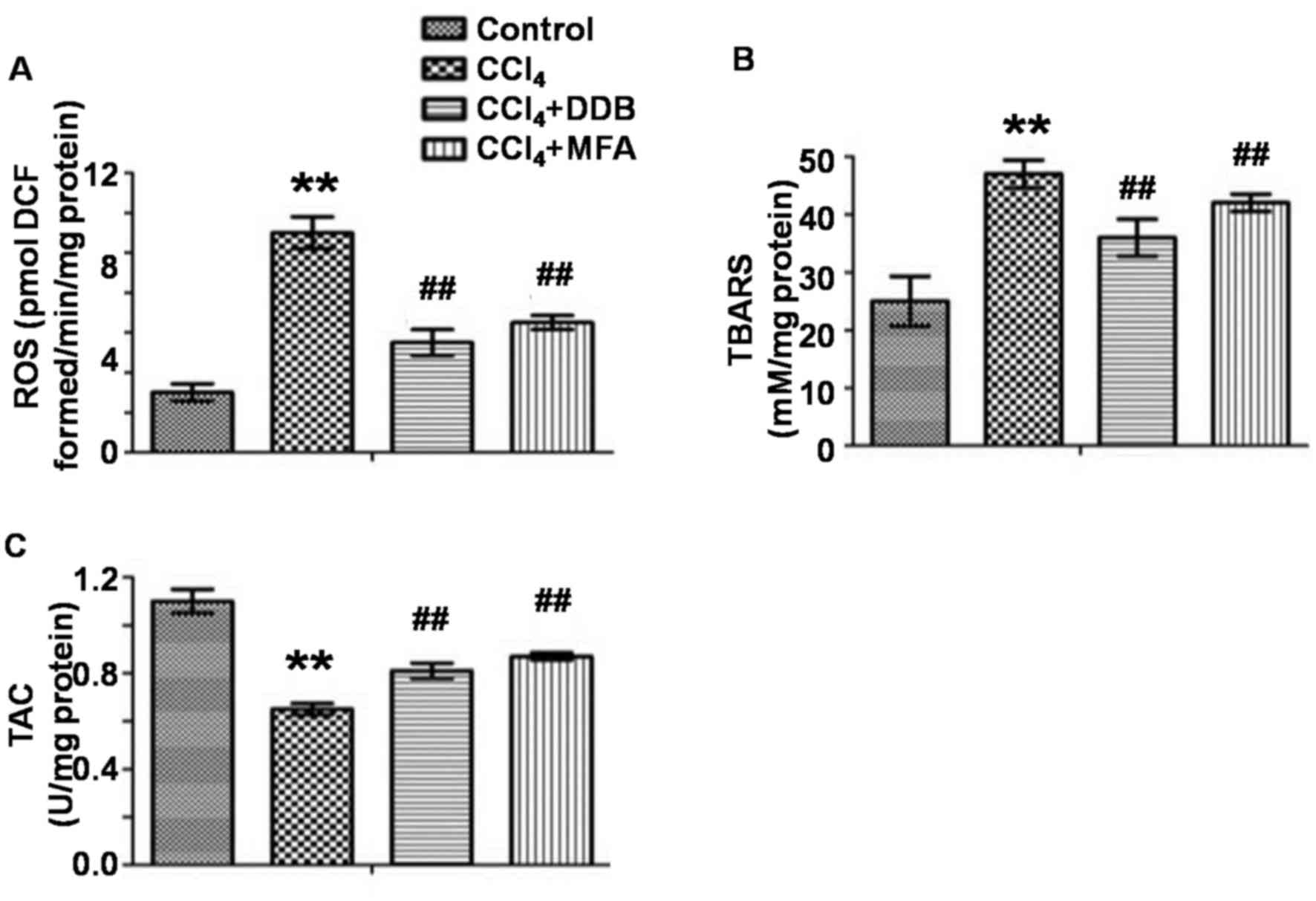
To evaluate oxidative stress in the liver, the
levels of ROS and TBARS as well as the TAC were measured. The
results demonstrated that CCl4 treatment markedly
improved hepatic ROS and TBARS levels, while decreasing the TAC
compared with those in the control group (P<0.01; Fig. 2A-C). Of note, pre-treatment with MFA
significantly reduced CCl4-induced ROS and TBARS
expression and significantly increased the TAC (P<0.01; Fig. 2A-C). These results indicated that MFA
inhibited oxidative stress induced by CCl4 in rat
livers.
MFA inhibits NOX4 and
p22phox mRNA and protein expression in the livers of
rats treated with CCl4
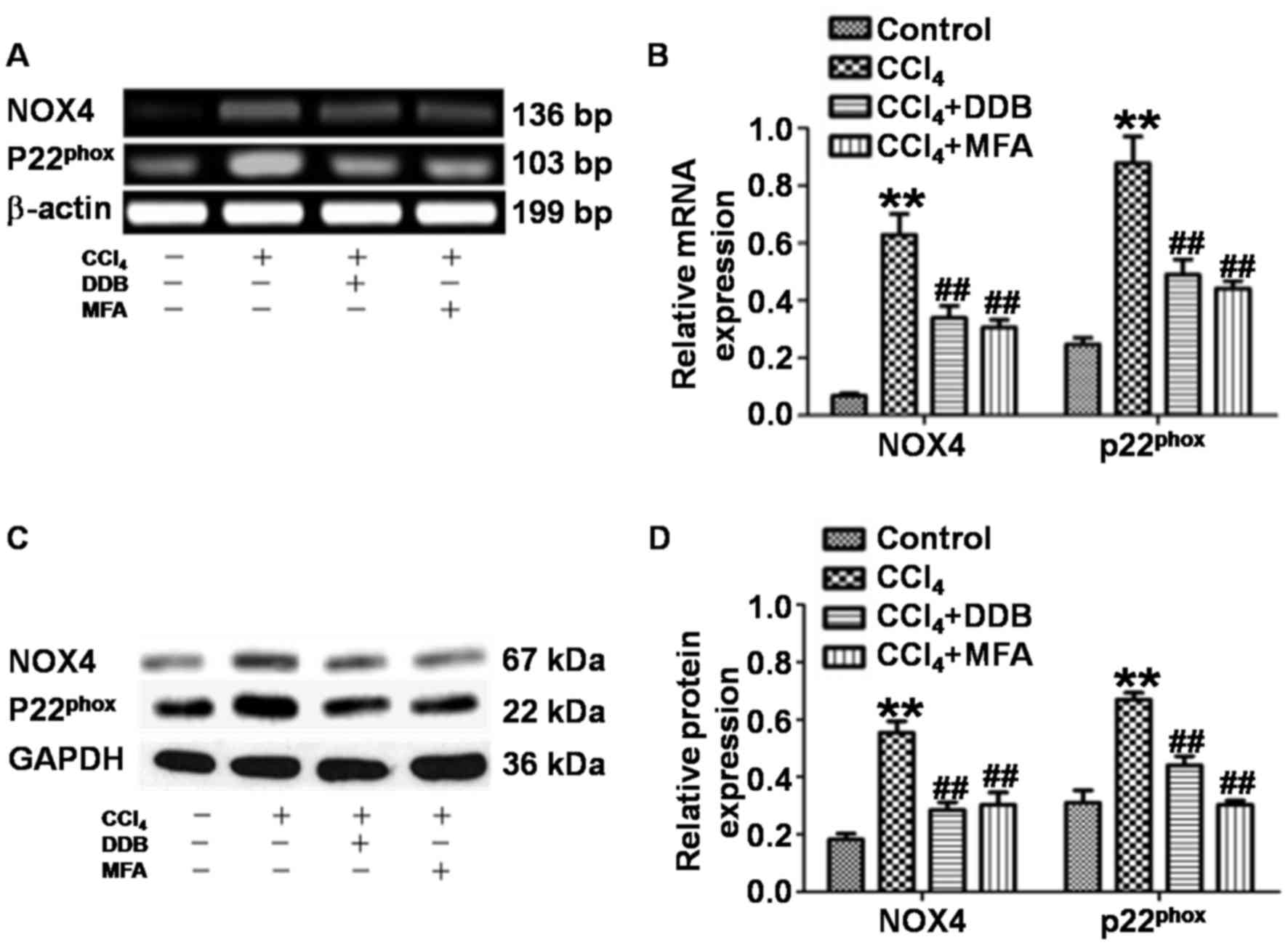
To assess whether MFA affects the generation of ROS
by inhibiting NOX4 and p22phox in acute liver injury
induced by CCl4, the present study determined the
expression of NOX4 and p22phox in liver tissues. qPCR
revealed that the levels of NOX4 and p22phox mRNA in the
livers of CCl4-treated rats were significantly increased
compared with those in the control group (P<0.01). By contrast,
pre-treatment with DDB or MFA (100 mg/kg) decreased the expression
of NOX4 and p22phox mRNA compared with that in rats
treated with CCl4 only (P<0.01; Fig. 3A and B). Western blot analysis
demonstrated significantly increased expression of NOX4 and
p22phox protein in the liver of CCl4-treated
rats compared with that in the control group (P<0.01). Of note,
the protein expression of NOX4 and p22phox was
significantly reduced by pre-treatment with DDB or MFA (100 mg/kg)
compared with that in rats treated with CCl4 only
(P<0.01; Fig. 3C and D). These
results suggested that pre-treatment with MFA inhibited the mRNA
and protein expression of NOX4 and p22phox in the livers
of rats treated with CCl4.
MFA mitigates CCl4-induced
pro-inflammatory responses by reducing the expression of TNF-α and
IL-1β
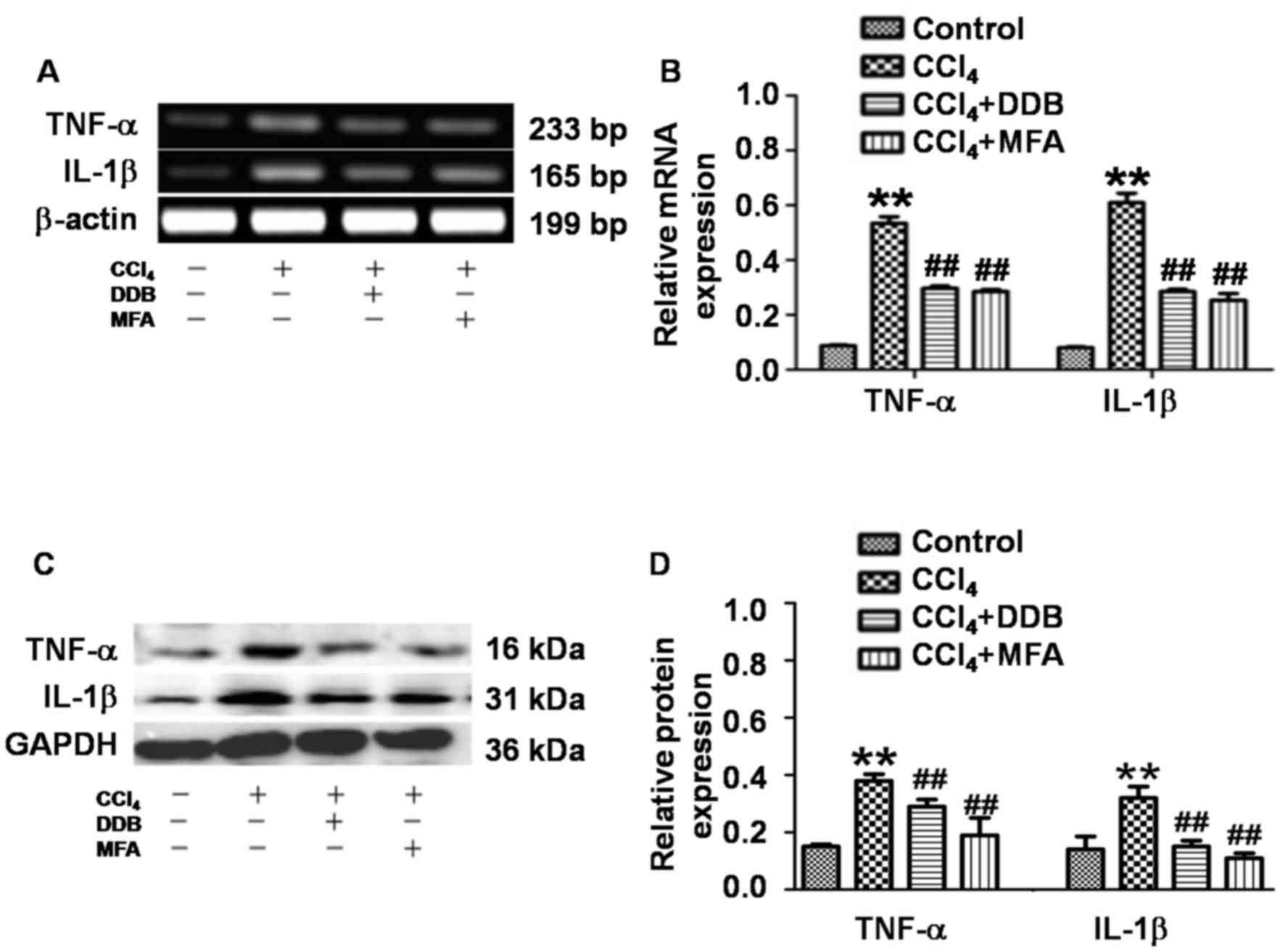
To determine the expression of TNF-α and IL-1β in
liver tissues, qPCR and western blot analysis were employed. The
results demonstrated that CCl4 treatment significantly
increased the hepatic TNF-α and IL-1β mRNA and protein expression
compared with that in the control group (P<0.01; Fig. 4A-D). Of note, pre-administration of
DDB or MFA significantly suppressed the CCl4-induced
mRNA and protein expression of hepatic TNF-α and IL-1β (P<0.01;
Fig. 4A-D). These results indicated
that pre-treatment with MFA prevented CCl4-induced
pro-inflammatory responses by inhibiting the expression of TNF-α
and IL-1β.
MFA inhibits CCl4-induced
apoptosis in the livers of rats
To investigate the effects of MFA on apoptosis
induced by CCl4, western blot analysis was used to
determine the ratio of Bax/Bcl-2 and the ratio of cleaved
caspase3/caspase3. The results demonstrated that CCl4
treatment markedly increased the expression of Bax compared with
that in the control group, while reducing the expression of Bcl-2,
leading to a significantly increased Bax/Bcl-2 ratio. However,
pre-treatment with MFA significantly decreased the
CCl4-induced expression of the pro-apoptotic protein Bax
and prominently decreased the Bax/Bcl-2 ratio as compared with that
in the CCl4 treatment group (P<0.01; Fig. 5A and B). In addition, cleaved
caspase3 levels in the livers of CCl4-treated rats were
significantly elevated as compared with those in the controls
(P<0.01). However, pre-treatment with MFA significantly
inhibited this CCl4-induced elevation (P<0.01;
Fig. 5C and D). These results
suggested that MFA inhibits CCl4-induced apoptosis in
the livers of rats.
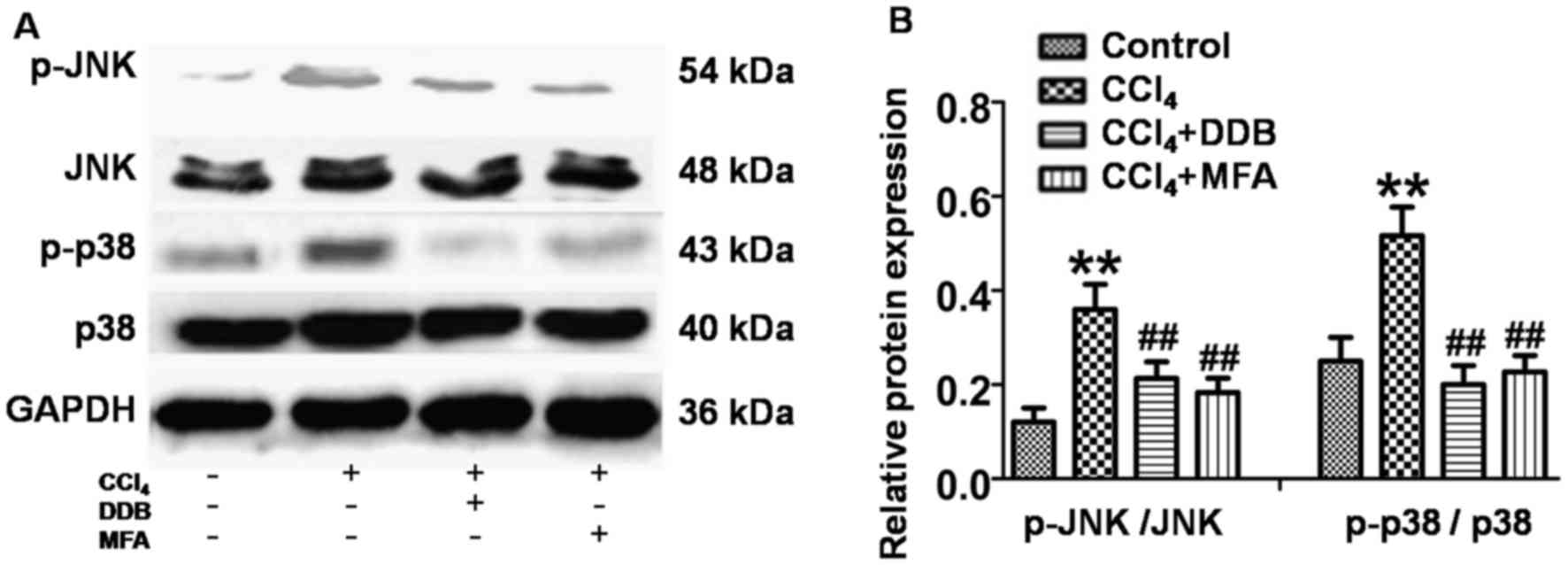
JNK and P38 MAPK activation is
involved in the anti-apoptotic effect of MFA
To investigate whether JNK and P38 MAPK signaling
was involved in the mechanism of action of MFA, the present study
investigated the effects of MFA on JNK and P38 MAPK in livers using
western blot analysis. The results revealed that the levels of
p-JNK and p-P38 MAPK were significantly increased in the livers of
CCl4-treated rats compared with those in the controls
(P<0.01; Fig. 6A and B). However,
this upregulation of p-JNK and p-p38 MAPK was significantly
suppressed by pre-treatment with MFA or DDP (P<0.01; Fig. 6A and B). These results indicated that
JNK and P38 MAPK activation is involved in the anti-apoptotic
effects of MFA.
Discussion
During liver injury, liver cells exhibit varying
degrees of swelling, degeneration, necrosis and apoptosis, which
are the most basic pathological states of the development of
various liver diseases. CCl4 has been widely used to
generate models of hepatic injury for evaluating plant-based drugs
for their hepatoprotective properties (3). It is well known that
CCl4-induced liver damage involves the formation of free
radicals (·CCl3) and the occurrence of lipid
peroxidation in cellular and organelle membranes (13). After entering the body,
CCl4 is metabolized by cytochrome P450 into free
radicals (·CCl3), which are mainly associated with
CCl4-induced hepatic damage. These free radicals react
with oxygen to form trichloromethylperoxy radicals
(CCl3OO·) and ROS, which trigger a chain reaction of
lipid peroxidation, and attack and destroy polyunsaturated fatty
acids, particularly those associated with phospholipids (27,28). All
of this results in the breakdown of cell integrity and leakage of
ALT and AST into the blood, leading to apoptosis and necrosis.
Overall, oxidative stress, caused by the overproduction of ROS, is
considered a vital risk factor in the development of liver disease.
Numerous studies suggested that the levels of ROS and TBARS as well
as the TAC may be indicators of oxidative stress (10,18). MFA
is a monomer isolated from Securidaca inappendiculata
Hasskarl with potent anti-viral activity (28). The present study investigated the
hepatoprotective activity of MFA using a rat model of
CCl4-induced acute liver damage, and DDB was used as a
positive control drug (29). The
results demonstrated that administration of MFA significantly
inhibited CCl4-induced elevation of serum ALT and AST
levels.
Oxidative stress has been postulated as an important
molecular mechanism in acute liver injury induced by
CCl4 (13,30). It was reported that the levels of MDA
and GSH-Px are associated with CCl4-induced, oxidative
stress-associated liver injury (29). MDA, the final product of lipid
peroxidation, gradually accumulates during CCl4-induced
liver injury and binds with biological macromolecules to form
aldehydes, further destroying cell membrane structure and function
(31). Increased MDA suggests
enhanced peroxidation that results in tissue damage and failure of
anti-oxidant defense mechanisms (32–34). The
results of the present study indicated that increased MDA during
CCl4-induced acute liver injury was prevented by
pre-treatment with MFA.
GSH is a main intracellular anti-oxidant that exerts
several main roles within a cell, including anti-oxidative effects,
maintenance of the redox state, detoxification of xenobiotics and
protection from damage by free radicals, toxins and peroxides
(35–37). It is well known that the depletion of
reduced GSH results in enhanced lipid peroxidation and
superabundant lipid peroxidation may cause increased GSH
consumption (38,39). Therefore, it is important to maintain
sufficient GSH levels for the prevention of CCl4-induced
damage. The results of the present study indicated that treatment
with MFA markedly inhibited the formation of MDA and increased the
level of GSH in the liver compared with that in the model group,
suggesting that MFA increases the anti-oxidant capacity, clears
free radicals and prevents cellular and organelle membranes from
being damaged by free radicals. MFA contains phenolic hydroxyl and
methoxy groups that directly or indirectly contribute to
anti-oxidant action (32,39).
As an effective metalloenzyme, SOD catalyzes the
dismutation of superoxide anions into hydrogen peroxide and
O2 (35). GSH-Px
catalyzes the reduction of toxic peroxide to a non-toxic hydroxyl
compound as well as the reduction of H2O2 and
hydroperoxides to water, removing lipid hydroperoxides from the
cell membrane to thereby terminate the chain reaction of lipid
peroxidation (32,35). The results of the present study
suggested that CCl4 treatment lowers the activities of
SOD and GSH-Px in the liver compared with those in the control
group. In addition, administration of MFA led to significantly
elevated activities of SOD and GSH-Px. Furthermore, MFA decreased
ROS and TBARS production in CCl4-treated livers due to
its powerful anti-oxidant and free radical scavenging activities.
In CCl4-induced liver injury, GSH has an important role
in detoxifying the toxic metabolites of CCl4, and once
GSH is exhausted, hepatocelluar necrosis or apoptosis begin
(40). In the present study, MFA
exerted hepatoprotective effects by reducing
CCl4-mediated oxidation of free radical species. In
addition, MFA attenuated hepatic glutathione depletion after
CCl4 treatment. In brief, the results of the present
study confirmed that MFA effectively reduced oxidative stress and
recovered anti-oxidant enzymes to their normal levels.
CCl4 has been reported to significantly
elevate the concentration of hydrogen peroxide and the amount of
lipid peroxidation in liver (41).
Studies suggested that overproduction of ROS has an important role
in the development and progression of CCl4-induced
hepatic damage (42–45). In line with this, the present study
also demonstrated the levels of ROS in CCl4-treated
model group were significantly higher than those in the control
group. Increased ROS generation stimulates pro-inflammatory
cytokines and results in oxidative damage to macromolecules. In
addition, pre-treatment with MFA significantly inhibited elevated
CCl4-induced decreases of SOD activity and increased MDA
levels in liver tissues of rats, suggesting that MFA exerts a
hepatoprotective effect partly through efficiently eliminating
excessive ROS in liver tissues. CCl4-induced ROS is
generated mainly through NOX-mediated pathways and it was reported
that NOX is a major source of ROS; furthermore, the NOX subunit
NOX4 and its ligand p22phox are highly expressed in
hepatocytes (30). NOX4 knock-out
rats exhibited lower hepatic lipid peroxidation after
CCl4 treatment compared with that in wild-type rats and
NOX4 deficiency was effective in preventing liver injury in rats
(46). The present study
demonstrated that MFA treatment for seven days decreased the levels
of ROS, NOX4 and its ligand p22phox. Overall, the
protective effect of MFA against acute liver injury may be partly
due to attenuating oxidative stress.
Apoptosis is a cell physiological self-extinction
process controlled by multiple genes (47,48). The
mitochondrial pathway is caused by a number of stress conditions,
chemical agents and drugs, and controlled by numerous genes. Bax
and Bcl-2 are important control factors (49), and caspase3 is the central effector
of apoptosis. Cleaved caspase3 may be used as a reliable indicator
to determine the severity of apoptosis (7,10,50). The
present study demonstrated that MFA caused upregulation of Bcl-2
expression and downregulation of the expression of Bax and cleaved
caspase3, leading to inhibition of apoptosis.
IL-1β and TNF-α are commonly considered as
biomarkers of inflammatory conditions. Serum IL-1β is markedly
increased during most inflammatory processes, and has been
demonstrated to prevent hepatocyte proliferation (51). TNF-α, which has an important role in
acute liver injury, is a mediator of hepatotoxicity (13). It activates intracellular pathways to
regulate inflammation and proliferation, and has been identified as
an attractive target for liver regeneration (13). TNF-α is also a pro-inflammatory
mediator in hepatocyte apoptosis, which is tightly associated with
cytotoxicity induced by CCl4 (13). In the present study, TNF-α and IL-1β
levels in liver tissues were significantly increased by
CCl4-induced hepatotoxicity, which was consistent with
the findings of a previous study (32). By contrast, TNF-α and IL-1β levels in
the MFA treatment group were lower than those in model group,
suggesting an anti-inflammatory role of MFA to prevent acute liver
injury.
The MAPK family is important for regulating cell
proliferation and death in response to various internal stresses.
JNKs are involved in stimulating apoptotic signaling. Oxidative
stress may activate JNK to cause apoptosis by receptor-initiated
extrinsic and mitochondrial intrinsic apoptotic pathways. JNKs also
has an essential role in modulating the functions of pro- and
anti-apoptotic proteins located in the mitochondria (12,52). JNK
and ROS stimulate the activities of pro-apoptotic proteins such as
Bax, and promote apoptosis by inhibiting anti-apoptotic proteins
such as Bcl-2 to regulate the release of cytochrome C and apoptosis
(10,52). The present study demonstrated that
the levels of p-JNK, TNF-α and Bax were increased in the livers of
CCl4-treated rats. Of note, pre-treatment with MFA
significantly repressed the CCl4-induced increases of
these proteins. Therefore, MFA exerted its protective effects on
the liver by regulating JNK signaling.
CCl4 was previously reported to
significantly increase the levels of p-p38 MAPK as a result of
oxidative stress and certain cytokines, leading to the activation
of p38 MAPK through its phosphorylation (15). The activation of the p38 MAPK pathway
accelerates cell apoptosis (16,17). In
the present study, the levels of p-p38/p38 MAPK ratio in the
CCl4-treated model group were higher than those in the
normal control group, which was consistent with the results of a
previous study (53). The present
study demonstrated that pre-treatment with MFA for seven days
decreased the levels of p-p38/p38 MAPK ratio, as well as ROS
levels. Therefore, the results demonstrated that during
CCl4 challenge, ROS produced by CCl4 promoted
the expression of p-p38 MAPK in the liver tissue, which in turn
resulted in necrosis and liver cell apoptosis. However, MFA
attenuated liver necrosis and cell apoptosis via reducing ROS
production. The histopathological observations of the present study
supported this notion.
p38 MAPK also causes mitochondria-dependent
apoptosis. p38 MAPK activation promotes mitochondrial translocation
of Bax and Bcl-2-like protein 11, while repressing the function of
Bcl-2 by increasing the phosphorylation of p38 MAPK, and induces
the activation of caspase3 (14).
Therefore, it is concluded that p38 MAPK and Bcl-2/Bax signaling
influence each other and cooperatively contribute to the protective
effect of MFA on the acute liver injury induced by
CCl4.
In summary, the present study demonstrated that MFA
had strong protective effects against CCl4-induced acute
oxidative liver injury and apoptosis by modulating JNK and p38 MAPK
as well as Bcl-2/Bax signaling pathways in the liver. MFA
alleviated CCl4-induced hepatic oxidative damage by
inhibiting ROS generation and increasing liver TAC. It also
effectively inhibited CCl4-induced inflammation and
apoptosis in the liver by upregulating p-JNK, p-P38 MAPK, Bax,
TNF-α and IL-1β, while downregulating Bcl-2 and cleaved caspase3.
These results provided evidence that MFA may be used as a
hepatoprotective agent for the treatment of liver diseases.
However, further study is necessary to fully elucidate the
molecular mechanisms that are responsible for the hepatoprotective
effects of MFA.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81360497).
References
|
1
|
Stickel F and Schuppan D: Herbal medicine
in the treatment of liver diseases. Dig Liver Dis. 39:293–304.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lal AA, Murthy PB and Pillai KS: Screening
of hepatoprotective effect of a herbal mixture against CCl4 induced
hepatotoxicity in Swiss albino mice. J Environ Biol. 28:201–207.
2007.PubMed/NCBI
|
|
3
|
Weber LW, Boll M and Stampfl A:
Hepatotoxicity and mechanism of action of haloalkanes: Carbon
tetrachloride as a toxicological model. Crit Rev Toxicol.
33:105–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berasain C, Castillo J, Perugorria MJ,
Latasa MU, Prieto J and Avila MA: Inflammation and liver cancer:
New molecular links. Ann N Y Acad Sci. 1155:206–221. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Campo GM, Avenoso A, Campo S, Nastasi G,
Traina P, D'Ascola A, Rugolo CA and Calatroni A: The antioxidant
activity of chondroitin-4-sulphate, in carbon tetrachloride-induced
acute hepatitis in mice, involves NF-kappaB and caspase activation.
Br J Pharmacol. 155:945–956. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang F, Wang X, Qiu X, Wang J, Fang H,
Wang Z, Sun Y and Xia Z: The protective effect of Esculentoside A
on experimental acute liver injury in mice. PLoS One.
9:e1131072014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Crosas-Molist E and Fabregat I: Role of
NADPH oxidases in the redox biology of liver fibrosis. Redox Biol.
6:106–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim EK and Choi EJ: Compromised MAPK
signaling in human diseases: An update. Arch Toxicol. 89:867–882.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma JQ, Ding J, Zhang L and Liu CM:
Hepatoprotective properties of sesamin against CCl4 induced
oxidative stress-mediated apoptosis in mice via JNK pathway. Food
Chem Toxicol. 64:41–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xie J, Liu J, Chen TM, Lan Q, Zhang QY,
Liu B, Dai D, Zhang WD, Hu LP and Zhu RZ: Dihydromyricetin
alleviates carbon tetrachloride-induced acute liver injury via
JNK-dependent mechanism in mice. World J Gastroenterol.
21:5473–5481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu Y, Hu D, Ma S, Zhao X, Wang S, Wei G,
Wang X, Wen A and Wang J: Protective effect of wedelolactone
against CCl4-induced acute liver injury in mice. Int
Immunopharmacol. 34:44–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Y, Wang R, Wang Y, Peng R, Wu Y and
Yuan Y: Ginkgo biloba extract mitigates liver fibrosis and
apoptosis by regulating p38 MAPK, NF-κB/IκBα and Bcl-2/Bax
signaling. Drug Des Devel Ther. 9:6303–6317. 2015.PubMed/NCBI
|
|
15
|
Bak J, Je NK, Chung HY, Yokozawa T, Yoon S
and Moon JO: Oligonol ameliorates CCl4-induced liver injury in rats
via the NF-Kappa B and MAPK signaling pathways. Oxid Med Cell
Longev. 2016:39358412016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ganai AA, Khan AA, Malik ZA and Farooqi H:
Genistein modulates the expression of NF-κB and MAPK (P-38 and
ERK1/2), thereby attenuating d-Galactosamine induced fulminant
hepatic failure in Wistar rats. Toxicol Appl Pharmacol.
283:139–146. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen S, Xuan J, Wan L, Lin H, Couch L, Mei
N, Dobrovolsky VN and Guo L: Sertraline, an antidepressant, induces
apoptosis in hepatic cells through the mitogen-activated protein
kinase pathway. Toxicol Sci. 137:404–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma JQ, Ding J, Zhang L and Liu CM: Ursolic
acid protects mouse liver against CCl4-induced oxidative stress and
inflammation by the MAPK/NF-κB pathway. Environ Toxicol Pharmacol.
37:975–983. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu CM, Zheng GH, Ming QL, Chao C and Sun
JM: Sesamin protects mouse liver against nickel-induced oxidative
DNA damage and apoptosis by the PI3K-Akt pathway. J Agric Food
Chem. 61:1146–1154. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng M: Inhibitory effect of 400 kinds of
Chinese herbal medicine on HBsAg. Chin J Integr Trad Western Med
Liver Dis. 1:341991.(In Chinese).
|
|
22
|
Qin Q, Yang X, Li Y, Li L, Li Y and Rong
M: Isolation and identification of extracting methyl ferulic acid
in cane peel onion. Asia Pac Trad Med. 10:20–21. 2014.
|
|
23
|
Li L, Li Y and Tang A: The inhibitory
effect of methyl ferulic acid on HBsAg and HBeAg in HepG2.2.15
cell. Pharmacol Clin Chin Mater Med. 27:14–16. 2011.(In
Chinese).
|
|
24
|
Li C, Li L, Yang CF, Zhong YJ, Wu D, Shi
L, Chen L and LI YW: Hepatoprotective effects of Methyl ferulic
acid on alcohol-induced liver oxidative injury in mice by
inhibiting the NOX4/ROS-MAPK pathway. Biochem Biophys Res Commun.
493:277–285. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta DeltaC(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li H, Sun JJ, Chen GY, Wang WW, Xie ZT,
Tang GF and Wei SD: Carnosic acid nanoparticles suppress liver
ischemia/reperfusion injury by inhibition of ROS, Caspases and
NF-κB signaling pathway in mice. Biomed Pharmacother. 82:237–246.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jain A, Soni M, Deb L, Jain A, Rout SP,
Gupta VB and Krishna KL: Antioxidant and hepatoprotective activity
of ethanolic and aqueous extracts of Momordica dioica Roxb. leaves.
J Ethnopharmacol. 115:61–66. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li L, Li Y, Tang A, Li M and Zhong Q: The
anti-inflammatory and immunopotentiation effect of chloroform
extracts isolated from Securidaca inappendiculata Hassk. Pharmacol
Clin Chin Mater Med. 27:62–64. 2011.(In Chinese).
|
|
29
|
Abdel-Hameid NA: Protective role of
dimethyl diphenyl bicarboxylate (DDB) against erythromycin induced
hepatotoxicity in male rats. Toxicol In Vitro. 21:618–625. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Roy S, Benz F, Alder J, Bantel H, Janssen
J, Vucur M, Gautheron J, Schneider A, Schüller F, Loosen S, et al:
Down-regulation of miR-192-5p protects from oxidative
stress-induced acute liver injury. Clin Sci (Lond). 130:1197–1207.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ismail AF, Salem AA and Eassawy MM:
Hepatoprotective effect of grape seed oil against carbon
tetrachloride induced oxidative stress in liver of γ-irradiated
rat. J Photochem Photobiol B. 160:1–10. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng N, Ren N, Gao H, Lei X, Zheng J and
Cao W: Antioxidant and hepatoprotective effects of Schisandra
chinensis pollen extract on CCl4-induced acute liver damage in
mice. Food Chem Toxicol. 55:234–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
NAIK SR and Panda VS: Antioxidants and
their role in biological functions: An overview. Indian Drugs.
27:393–399. 2007.
|
|
34
|
Pareek A, Godavarthi A, Issarani R and
Nagori BP: Antioxidant and hepatoprotective activity of Fagonia
schweinfurthii (Hadidi) Hadidi extract in carbon tetrachloride
induced hepatotoxicity in HepG2 cell line and rats. J
Ethnopharmacol. 150:973–981. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ai G, Liu Q, Hua W, Huang Z and Wang D:
Hepatoprotective evaluation of the total flavonoids extracted from
flowers of Abelmoschus manihot (L.) Medic: In vitro and in vivo
studies. J Ethnopharmacol. 146:794–802. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yuan L and Kaplowitz N: Glutathione in
liver diseases and hepatotoxicity. Mol Aspects Med. 30:29–41. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Townsend DM, Tew KD and Tapiero H: The
importance of glutathione in human disease. Biomed Pharmacother.
57:145–155. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dong Y, Huang J, Lin X, Zhang S, Jiao Y,
Liang T, Chen Z and Huang R: Hepatoprotective effects of Yulangsan
polysaccharide against isoniazid and rifampicin-induced liver
injury in mice. J Ethnopharmacol. 152:201–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Onyema OO, Farombi EO, Emerole GO, Ukoha
AI and Onyeze GO: Effect of vitamin E on monosodium glutamate
induced hepatotoxicity and oxidative stress in rats. Indian J
Biochem Biophys. 43:20–24. 2006.PubMed/NCBI
|
|
40
|
Deng JS, Chang YC, Wen CL, Liao JC, Hou
WC, Amagaya S, Huang SS and Huang GJ: Hepatoprotective effect of
the ethanol extract of Vitis thunbergii on carbon
tetrachloride-induced acute hepatotoxicity in rats through
anti-oxidative activities. J Ethnopharmacol. 142:795–803. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiang M, Wang J, Zhang Y, Ling J and Xu X:
Attenuation of aortic injury by ursolic acid through RAGE-Nox-NFκB
pathway in streptozocin-induced diabetic rats. Arch Pharm Res.
35:877–886. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ranawat L, Bhatt J and Patel J:
Hepatoprotective activity of ethanolic extracts of bark of
Zanthoxylum armatum DC in CCl4 induced hepatic damage in rats. J
Ethnopharmacol. 127:777–780. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu D, Zhai Q and Shi X: Alcohol-induced
oxidative stress and cell responses. J Gastroenterol Hepatol. 21
Suppl 3:S26–S29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Stehbens WE: Oxidative stress, toxic
hepatitis, and antioxidants with particular emphasis on zinc. Exp
Mol Pathol. 75:265–276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li J, Pan Y, Kan M, Xiao X, Wang Y, Guan
F, Zhang X and Chen L: Hepatoprotective effects of berberine on
liver fibrosis via activation of AMP-activated protein kinase. Life
Sci. 98:24–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lan T, Kisseleva T and Brenner DA:
Deficiency of NOX1 or NOX4 prevents liver inflammation and fibrosis
in mice through inhibition of hepatic stellate cell activation.
PLoS One. 10:e01297432015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Green DR and Fitzgerald P: Just So stories
about the evolution of apoptosis. Curr Biol. 26:R620–R627. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Savitskaya MA and Onishchenko GE:
Mechanisms of apoptosis. Biochemistry (Mosc). 80:1393–1405. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lindsay J, Esposti MD and Gilmore AP:
Bcl-2 proteins and mitochondria-specificity in membrane targeting
for death. Biochim Biophys Acta. 1813:532–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang W, Yin L, Tao X, Xu L, Zheng L, Han
X, Xu Y, Wang C and Peng J: Dioscin alleviates
dimethylnitrosamine-induced acute liver injury through regulating
apoptosis, oxidative stress and inflammation. Environ Toxicol
Pharmacol. 45:193–201. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tien YC, Liao JC, Chiu CS, Huang TH, Huang
CY, Chang WT and Peng WH: Esculetin ameliorates carbon
tetrachloride-mediated hepatic apoptosis in rats. Int J Mol Sci.
12:4053–4067. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kim HY, Park J, Lee KH, Lee DU, Kwak JH,
Kim YS and Lee SM: Ferulic acid protects against carbon
tetrachloride-induced liver injury in mice. Toxicology.
282:104–111. 2011. View Article : Google Scholar : PubMed/NCBI
|