Introduction
Oxidative stress injury refers to tissue and cell
damage due to an imbalance between oxidation and anti-oxidation in
the body when exposed to a variety of internal and external stimuli
(1). Oxidative stress is considered
the main cause of various cardiovascular diseases such as
myocardial reperfusion injury, myocardial infarction and
atherosclerosis (2). A large amount
of reactive oxygen species (ROS) is generated in the process of
myocardial ischemia-reperfusion, resulting in the apoptosis of
myocardial cells due to oxidative damage. Therefore, oxidative
stress is the main culprit of myocardial cell death in myocardial
ischemia-reperfusion (2,3).
Hydrogen peroxide (H2O2) is a
molecule belonging to ROS that can easily pass through the cell
membrane. It converts into other more reactive ROS in the body,
which directly oxidize the lipids and proteins on the cell
membrane, resulting in apoptosis through multiple pathways
(4). At present,
H2O2-induced oxidative stress injury of
myocardial cells has become a commonly used in vitro
experimental model of myocardial ischemia-reperfusion injury
(5). Endoplasmic reticulum stress is
an imbalance stress process in cell endoplasmic reticulum, which
leads to physiological dysfunction (6). It was found that endoplasmic reticulum
stress plays a key role in multiple disease processes such as
myocardial ischemia-reperfusion injury and myocardial infarction
(7).
In clinical applications, sevoflurane was found to
exert protective effect on myocardial cells. For example,
sevoflurane treatment played a role in enhancing myocardial
contractility (6), slowing heart
rate (7), lowering left ventricular
diastolic pressure (8), and reducing
ischemia-reperfusion injury to myocardial cells (9). It was reported that the mechanism
underlying inhibition of myocardial cell apoptosis through
sevoflurane treatment may be due to the upregulated expression of
anti-apoptotic Bcl-2 protein, downregulated expression of
pro-apoptotic Bax protein, and resulted inhibition of the
mitochondrial apoptotic signaling pathways (10). At present there is no study on
whether sevoflurane can reduce endoplasmic reticulum stress to
inhibit oxidative stress-induced cardiomyocyte apoptosis. In this
study, rat H9c2 cardiomyocytes were used to establish an in
vitro experimental model of H2O2-induced
oxidative stress to examine the inhibitory effect of sevoflurane
pretreatment on H2O2-induced cardiomyocyte
injury and apoptosis. In particular, the expression levels of
glucose-regulated protein 78 (GRP78) mRNA and protein as stress
indicators in endoplasmic reticulum, the intracellular free
Ca2+ concentration, and the downstream protein
expression levels of CHOP and caspase-12 in endoplasmic reticulum
stress were measured, in order to investigate whether
sevoflurane-mediated inhibition of
H2O2-induced cardiomyocyte apoptosis was
associated with the reduction of endoplasmic reticulum stress.
Materials and methods
Materials
The following materials were purchased from
commercial sources: Rat embryonic ventricular myocardium H9c2 from
the Chinese Academy of Sciences Cell Bank (Beijing, China);
Dulbeccos modified Eagles medium (DMEM) medium and fetal bovine
serum (FBS) from HyClone Laboratories (Logan, UT, USA); GRP78,
CHOP, caspase-12, glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
primary antibody, and HRP secondary antibody from Proteintech
Group, Inc. (Wuhan, China); BCA protein concentration assay kit,
Annexin V-FITC apoptosis detection kit, and Fluo-3 AM calcium ion
detection kit from Beyotime Biotechnology, Inc. (Nantong, China);
TRIzol kit, reverse transcription kit, and RT-PCR kit from
Invitrogen (Carlsbad, CA, USA); primer synthesis from Takara
(Dalian, China); hydrogen peroxide, sevoflurane, and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
from Sigma (St. Louis, MO, USA).
Cell culture
H9c2 cells were cultured in DMEM supplemented with
10% fetal bovine serum at 37°C in a humidified incubator containing
5% CO2. When cells covered 80% of the bottom of the
culture container, the cells were treated with trypsin to obtain a
single cell suspension, and then seeded into different culture
plates for group experiments according to the experimental
requirement.
Viability of H9c2 cells determined by
MTT assay
H9c2 cells in the logarithmic growth phase were
collected and seeded into 96-well plates. After 24 h incubation,
the cells were divided into the control, model and sevoflurane
groups. The cells in the control group received no treatment, cells
in the model group were treated with 400 µM
H2O2, and cells in the sevoflurane group were
pretreated with sevoflurane followed by treatment with 400 µM
H2O2. The sevoflurane pretreatment was
performed as previously mentioned (11). H9c2 cells were placed in a sterile
sealed container connected with the breathing circuit of an
anesthesia machine. The sevoflurane vaporizer was filled with
sevoflurane and the vaporizer dial was set at 2.5%. The
concentration of sevoflurane in the outlet of the closed container
was monitored. When it reached 2.5%, the sevoflurane flow was
maintained for 20 min, after which the cells were returned to a
normal incubator for another 10 min (11). The hydrogen peroxide treatment was
carried out by incubation of the cells with a final concentration
of 400 µM H2O2 to generate the
H2O2 stress injury model (12). After 4 h incubation with
H2O2, the culture medium was discarded, and
the wells were washed three times with phosphate-buffered saline
(PBS), followed by the addition of 100 µl MTT solution (5 mg/ml)
per well and incubation for 4 h. Then, 100 µl dimethyl sulfoxide
(DMSO) was added to each well, followed by agitation for 10 min and
measurement of absorbance value at 570 nm using a microplate reader
(model 680; Bio-Rad Laboratories, Hercules, CA, USA). The cell
survival rate was calculated using the formula: Survival rate (%) =
100 × (OD value of experimental group/OD value of control
group).
Apoptosis of H9c2 cells determined by
Annexin V-propidium iodide (AV-PI) double staining assay
H9c2 cells in the logarithmic phase were collected
and seeded into 6-well plates. The cells were grouped and processed
following the protocol described above. After
H2O2 treatment, the cells were treated with
0.25% trypsin, followed by three washes with PBS. The cell
concentration was then adjusted to 5×105 cells/ml by the
addition of 300 µl binding buffer. Then, 10 µl Annexin V-FITC and 5
µl PI were added to each sample well. After incubation for 15 min
at room temperature in the dark, the samples were diluted with
another 300 µl binding buffer, followed by filtration through a 300
mesh cell strainer. The apoptotic rate of each sample was measured
by flow cytometry (Becton-Dickinson and Company, Franklin Lakes,
NJ, USA).
Intracellular Ca2+ levels
of H9c2 cells measured by fluorescence-based calcium ion probe
using Fluo-3 AM
H9c2 cells in the logarithmic phase were collected
and seeded into 6-well plates. The cells were grouped and processed
following the protocol described above. After treatment with
H2O2, the cells were digested with trypsin
followed by centrifugation at 8,000 × g for 10 min. The remaining
steps were performed following the instructions of the calcium
assay kit. The collected cells were re-suspended in 500 µl staining
buffer containing 2.5 µM Fluo-3 AM. After incubation at 37°C for 40
min, the samples were analyzed by flow cytometry (Becton-Dickinson
and Company).
Expression levels of GRP78 mRNA in
H9c2 cells measured by RT-qPCR
H9c2 cells in the logarithmic phase were collected
and seeded into 6-well plates. The cells were grouped and processed
following the protocol described above. After treatment with
H2O2, the cells were digested with trypsin,
followed by centrifugation 8,000 × g. Total RNA was extracted from
the collected cells in each group with TRIzol reagent. Then cDNA
was synthesized from the RNA template via reverse transcription
using oligo(dT) primers. PCR amplification was carried out on the
obtained cDNA using the primers listed in Table I. PCR conditions were as follows:
95°C for 10 min; then 40 cycles of 95°C for 15 sec and 60°C for 1
min. The GAPDH gene was used as the reference gene. Cq
values were obtained from the instrument outputs. Relative
quantification of gene expression between different groups was
estimated by calculation of 2−ΔCq, where ΔCqtarget
gene = Cqtarget gene-Cqreference
gene.
 | Table I.RT-qPCR primer sequences. |
Table I.
RT-qPCR primer sequences.
| Genes | Primer
sequences |
|---|
| GRP78 | F:
5′-CTGGGTACATTTGATCTGACTGG-3′ |
|
| R:
5′-GCATCCTGGTGGCTTTCCAGCCATTC-3′ |
| GAPDH | F:
5′-GCACCGTCAAGGCTGAGAAC-3′ |
|
| R:
5′-TGGTGAAGACGCCAGTGGA-3′ |
Expression levels of proteins GRP78,
CHOP and caspase-12 measured by western blot analysis
H9c2 cells in the logarithmic phase were collected
and seeded into 6-well plates. The cells were grouped and processed
following the protocol described above. After treatment with
H2O2, the cells were digested with trypsin,
followed by centrifugation. The collected cells were lysed using
RIPA cell lysis buffer. Proteins were extracted, and the
concentrations were measured using BCA assay. A sample containing
40 µg of proteins from each group was loaded onto a 12% gel for
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) analysis. The protein bands were transferred to PVDF
membrane using a wet system. After the membrane was blocked with 5%
skimmed milk powder for 2 h, the primary antibodies of GRP78, CHOP,
caspase-12 and GAPDH (dilution, 1:1,000; cat. nos. 11587-1-AP,
15204-1-AP, 55238-1-AP and 10494-1-AP) were added, respectively,
followed by incubation overnight at 4°C. After the membrane was
washed three times with TBST buffer, the HRP-labeled secondary
antibody (1:1,000 dilution) was added, followed by incubation at
room temperature for 2 h. The ECL reagent was applied to the
membrane in the dark after it was washed three times with TBST. The
image of the membrane was scanned with a digital imager and was
used in statistical analysis.
Statistical analysis. The experimental data were
expressed as mean ± standard deviation, and were processed using
SPSS 13.0 software (International Business Machines Corp., New
York, NY, USA). Data were analyzed with single factor analysis of
variance, and t-test was used in paired comparison. Differences
were statistically significant at P<0.05.
Results
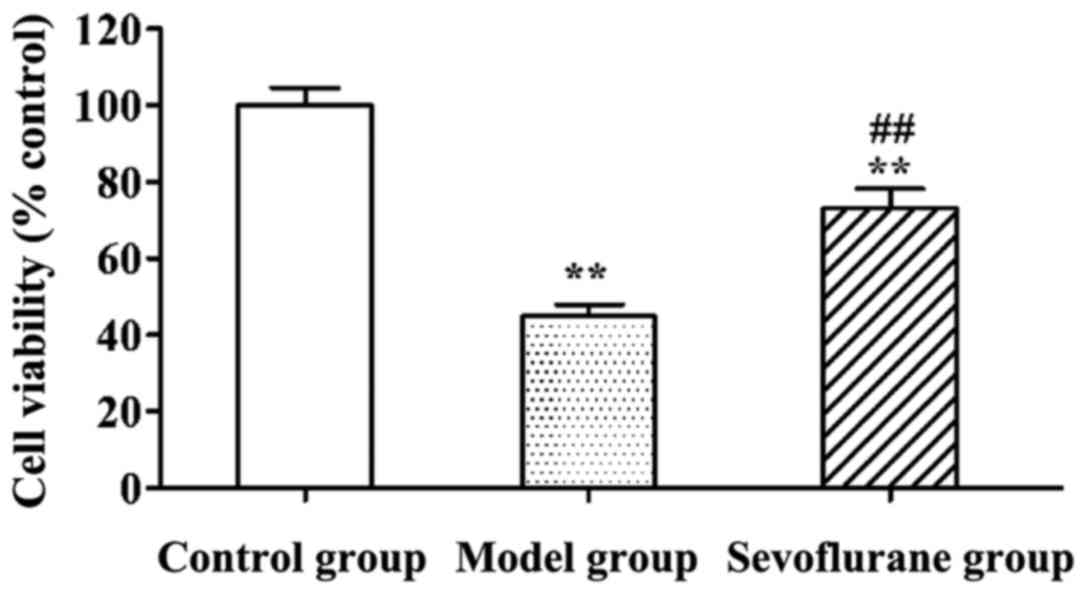
Effect of sevoflurane on viabilities
of H9c2 cells treated with H2O2
Viabilities of H2O2-treated
H9c2 cells determined using MTT assay are shown in Fig. 1. Viabilities of H9c2 cells in the
model and sevoflurane groups were significantly lower than that in
the control group (P<0.01). Moreover, the viability in the
sevoflurane-pretreated group was higher than that in the model
group (P<0.01).
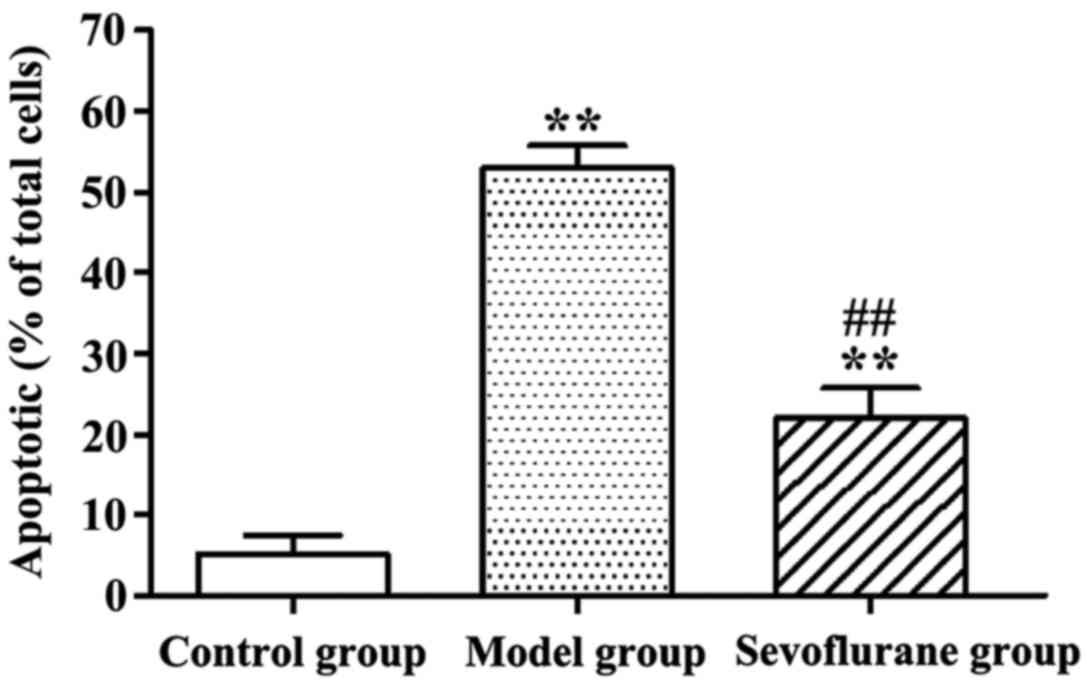
Effect of sevoflurane on
H2O2-induced apoptosis of H9c2 cells
Apoptotic rates of
H2O2-treated H9c2 cells determined by AV-PI
double staining assay are shown in Fig.
2. Evidently, after H9c2 cells were treated with
H2O2, the apoptotic rate in the model group
was significantly increased as compared with that in the control
group (P<0.01). By contrast, cells pretreated with sevoflurane
prior to H2O2 challenge, showed an increased
apoptotic rate (sevoflurane group), but which was much lower than
that in the model group (P<0.01).
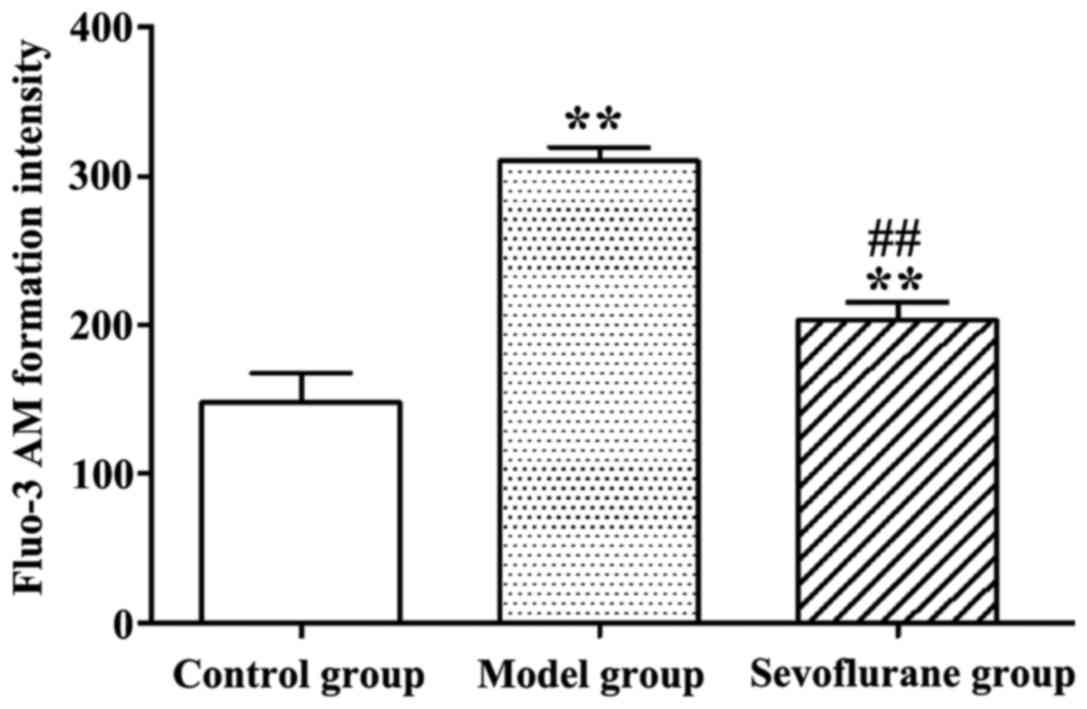
Effect of sevoflurane on intracellular
Ca2+ concentration in H2O2-treated
H9c2 cells
As shown in Fig. 3,
after H9c2 cells were treated with H2O2, the
intracellular Ca2+ concentration in the model group was
significantly increased (P<0.01), compared to the normal value
in the control group. By contrast, when cells were pretreated with
sevoflurane prior to H2O2 challenge, the
intracellular Ca2+ concentration as seen in the
sevoflurane group, was higher than that in the control group, but
much lower than that in the model group (P<0.01). This result
suggested that sevoflurane pretreatment could balance the increase
of the intracellular Ca2+ concentration induced by
H2O2.
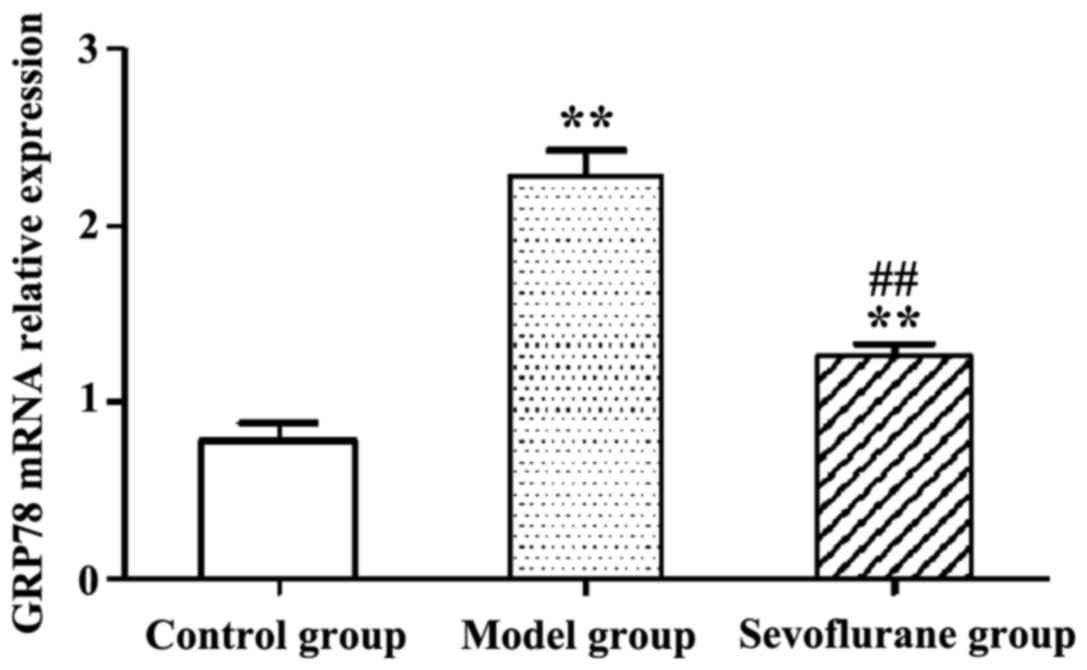
Effect of sevoflurane on the
expression levels of GRP78 mRNA in
H2O2-treated H9c2 cells
RT-qPCR results show that following
H2O2-treatment, the expression level of
intracellular GRP78 mRNA in the model group was significantly
increased (P<0.01) (Fig. 4). By
contrast then the cells were pretreated with sevoflurane prior to
the H2O2 challenge, the expression level of
GRP78 mRNA as shown in the sevoflurane group was increased, but
much lower than that in the model group (P<0.01). This result
suggested that pretreatment with sevoflurane could balance the
increase of intracellular GRP78 mRNA expression induced by
H2O2.
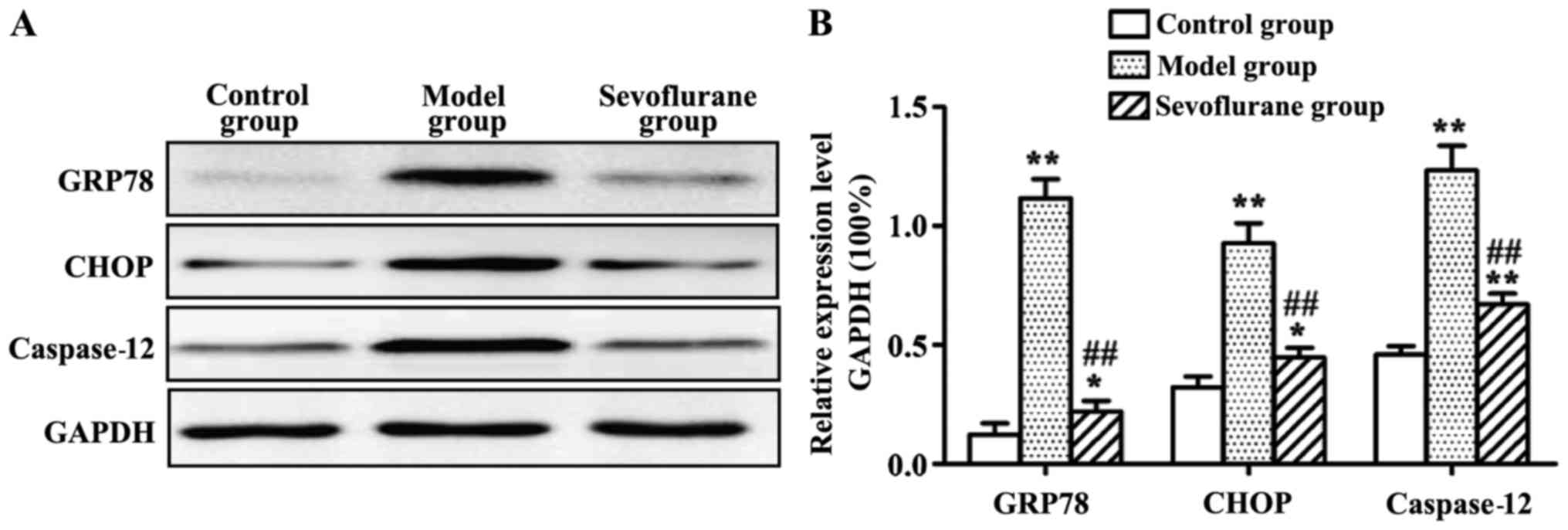
Effect of sevoflurane on the protein
expression levels of GRP78, CHOP and caspase-12 in
H2O2-induced H9c2 cells
Protein expression levels of GRP78, CHOP and
caspase-12 measured by western blot analysis are shown in Fig. 5. The results showed that, after
H2O2-treatment the intracellular protein
expression levels of GRP78, CHOP and caspase-12 in the model group
were all substantially increased (P<0.01). By contrast, when the
cells were pretreated with sevoflurane prior to
H2O2 challenge, the three protein levels were
increased as shown in the sevoflurane group, but much lower than
those in the model group (P<0.01).
Discussion
Recent findings have shown that oxidative stress was
closely associated with the onset and progression of cardiovascular
diseases. In the process of myocardial ischemia-reperfusion, a
large amount of ROS were produced, and the antioxidant capacity of
tissues and cells became weak as well, leading to oxidative stress
response that caused myocardial apoptosis (13,14).
Hydrogen peroxide (H2O2) belongs to ROS and
is a common molecule. It modulates the activity of transcription
factor and related gene expression in the body. Excessive
H2O2 can induce apoptosis (15–17).
According to the literature, currently known mechanisms underlying
myocardial apoptosis mainly include the death receptor,
mitochondrial and endoplasmic reticulum pathways. Due to rich
endoplasmic reticulum in cardiomyocytes, the endoplasmic reticulum
pathway may play a key role in cardiomyocyte apoptosis (3).
GRP78 is an important molecular chaperone in
endoplasmic reticulum, which modulates endoplasmic reticulum stress
by maintaining correct synthesis and folding of endoplasmic
reticulum proteins and stabilizing intracellular calcium
concentration (18). It was found
that the GRP78 protein expression level in the stress state can be
increased several times to cope with modulation of the accumulation
and release of calcium in the endoplasmic reticulum, thus
maintaining calcium balance in the endoplasmic reticulum (19). Intracellular calcium overload is one
of the major causes of myocardial ischemia-reperfusion injury.
Prevention of intracellular calcium overload has become an
important target for the treatment of myocardial
ischemia-reperfusion injury (20).
CHOP is induced by endoplasmic reticulum stress and mediates
apoptosis. It plays a key role in cell proliferation and apoptosis
(21). CHOP is downstream of the
apoptotic pathway induced by endoplasmic reticulum stress.
Therefore, the expression level of CHOP can reflect the degree of
apoptosis induced by endoplasmic reticulum stress (22). In addition, caspase-12 located in the
endoplasmic reticulum membrane plays an important role in
endoplasmic reticulum stress-induced apoptosis (23).
In this study, rat H9c2 cardiomyocytes were employed
to establish a model of oxidative stress injury induced by
H2O2. The model was used to investigate the
effect of sevoflurane pretreatment on myocardial injury and
apoptosis induced by H2O2. Viabilities of
H2O2-treated H9c2 cells were determined using
MTT assay. The results indicated that viability of H9c2 cells in
the model and sevoflurane groups was significantly lower than that
in the control group. Moreover, the viability in the
sevoflurane-pretreated group was higher than that in the model
group. This suggested that sevoflurane had a protective effect on
H9c2 cardiomyocytes from H2O2-induced injury.
The results of AV-PI double staining assay showed that pretreatment
with sevoflurane could reverse H2O2-induced
apoptosis of H9c2 cells. After H9c2 cells were treated with
H2O2, the intracellular Ca2+
concentration in the model group was significantly increased,
compared to the normal value in the control group. By contrast when
cells were pretreated with sevoflurane prior to
H2O2 challenge, the intracellular
Ca2+ concentration as seen in the sevoflurane group was
higher than that in the control group, but much lower than that in
the model group. This result suggests that sevoflurane pretreatment
can balance the increase of the intracellular Ca2+
concentration induced by H2O2. The RT-qPCR
results suggest that sevoflurane pretreatment can balance the
increase of intracellular GRP78 mRNA expression induced by
H2O2. Western blot analysis revealed that
H2O2 induced the expression of GRP78 and the
downstream proteins CHOP and caspase-12 in the endoplasmic
reticulum stress. By contrast, when the cells were pretreated with
sevoflurane prior to H2O2 challenge,
expression levels of the three proteins were increased as compared
with the control group, but much lower than those in the model
group.
It was reported that sevoflurane pretreatment
reduced myocardial injury caused by myocardial
ischemia-reperfusion. This effect of sevoflurane was probably
associated with the inhibition of cardiomyocyte apoptosis, but the
mechanism was not clear (11,24).
Myocardial ischemia-reperfusion induced overexpression of
intracellular GRP78 protein, triggering the endoplasmic reticulum
stress response and increase the expression of CHOP protein, the
phosphorylation of JNK and activation of caspase-12. The three
proteins were involved in endoplasmic reticulum stress-induced
myocardial apoptosis (25). Thus,
the results not only confirmed that sevoflurane reduced the injury
and apoptosis of H9c2 cardiomyocytes induced by
H2O2, but also confirmed that sevoflurane
downregulated the expression of GRP78 mRNA and that of proteins
GRP78, CHOP and caspase-12 as well.
In conclusion, results of this study have shown that
sevoflurane can reduce H2O2-induced injury
and apoptosis of H9c2 cardiomyocytes. The mechanism may be related
to inhibition of the expression of GRP78, a regulating protein for
endoplasmic reticulum stress, thereby reducing intracellular
Ca2+ concentration, and the downregulation of CHOP and
caspase-12 expression levels.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZW analyzed and interpreted the patient data, and
was a major contributor in writing the manuscript. RC performed the
experiment and participated in the design of the study. KW was a
major contributor in designing the methods.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
People's Hospital of Rizhao.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Delbosc S, Paizanis E, Magous R, Araiz C,
Dimo T, Cristol JP, Cros G and Azay J: Involvement of oxidative
stress and NADPH oxidase activation in the development of
cardiovascular complications in a model of insulin resistance, the
fructose-fed rat. Atherosclerosis. 179:43–49. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang GG, Teng X, Liu Y, Cai Y, Zhou YB,
Duan XH, Song JQ, Shi Y, Tang CS, Yin XH, et al: Inhibition of
endoplasm reticulum stress by ghrelin protects against
ischemia/reperfusion injury in rat heart. Peptides. 30:1109–1116.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gustafsson AB and Gottlieb RA: Mechanisms
of apoptosis in the heart. J Clin Immunol. 23:447–459. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sharikabad MN, Østbye KM and Brørs O:
Effect of hydrogen peroxide on reoxygenation-induced
Ca2+ accumulation in rat cardiomyocytes. Free Radic Biol
Med. 37:531–538. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Benvenga S, Vicchio T, Di Bari F, Vita R,
Fallahi P, Ferrari SM, Catania S, Costa C and Antonelli A:
Favorable effects of myo-inositol, selenomethionine or their
combination on the hydrogen peroxide-induced oxidative stress of
peripheral mononuclear cells from patients with Hashimoto's
thyroiditis: Preliminary in vitro studies. Eur Rev Med Pharmacol
Sci. 21 Suppl 2:89–101. 2017.PubMed/NCBI
|
|
6
|
Schröder M: Endoplasmic reticulum stress
responses. Cell Mol Life Sci. 65:862–894. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Glembotski CC: Endoplasmic reticulum
stress in the heart. Circ Res. 101:975–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bouwman RA, Salic K, Padding FG, Eringa
EC, van Beek-Harmsen BJ, Matsuda T, Baba A, Musters RJ, Paulus WJ,
de Lange JJ, et al: Cardioprotection via activation of protein
kinase C-delta depends on modulation of the reverse mode of the
Na+/Ca2+ exchanger. Circulation. 114(Suppl
1): I226–I232. 2006.PubMed/NCBI
|
|
9
|
Yildirim V, Doganci S, Aydin A, Bolcal C,
Demirkilic U and Cosar A: Cardioprotective effects of sevoflurane,
isoflurane, and propofol in coronary surgery patients: A randomized
controlled study. Heart Surg Forum. 12:E1–E9. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Inamura Y, Miyamae M, Sugioka S, Domae N
and Kotani J: Sevoflurane postconditioning prevents activation of
caspase 3 and 9 through antiapoptotic signaling after myocardial
ischemia-reperfusion. J Anesth. 24:215–224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Allaouchiche B, Debon R, Goudable J,
Chassard D and Duflo F: Oxidative stress status during exposure to
propofol, sevoflurane and desflurane. Anesth Analg. 93:981–985.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eguchi M, Liu Y, Shin EJ and Sweeney G:
Leptin protects H9c2 rat cardiomyocytes from
H2O2-induced apoptosis. FEBS J.
275:3136–3144. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee Y and Gustafsson AB: Role of apoptosis
in cardiovascular disease. Apoptosis. 14:536–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
von Harsdorf R, Li PF and Dietz R:
Signaling pathways in reactive oxygen species-induced cardiomyocyte
apoptosis. Circulation. 99:2934–2941. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ricci F, Masini F, Fossati B, Frascione P,
De Waure C, Capizzi R and Guerriero C: Combination therapy with
hydrogen peroxide (4%), salicylic acid (0.5%) and D-panthenol (4%):
Efficacy and skin tolerability in common acne vulgaris during sun
exposure period. Eur Rev Med Pharmacol Sci. 20:232–236.
2016.PubMed/NCBI
|
|
16
|
Then SM, Sanfeliu C, Top GM, Wan Ngah WZ
and Mazlan M: γ-Tocotrienol does not substantially protect DS
neurons from hydrogen peroxide-induced oxidative injury. Nutr Metab
(Lond). 9:12012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fu J, Huang H, Liu J, Pi R, Chen J and Liu
P: Tanshinone IIA protects cardiac myocytes against oxidative
stress-triggered damage and apoptosis. Eur J Pharmacol.
568:213–221. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shu CW, Sun FC, Cho JH, Lin CC, Liu PF,
Chen PY, Chang MD, Fu HW and Lai YK: GRP78 and Raf-1 cooperatively
confer resistance to endoplasmic reticulum stress-induced
apoptosis. J Cell Physiol. 215:627–635. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang YM, Yang Y, Dai WW, Li XM, Ma JQ and
Tang LP: Genistein-induced apoptosis is mediated by endoplasmic
reticulum stress in cervical cancer cells. Eur Rev Med Pharmacol
Sci. 20:3292–3296. 2016.PubMed/NCBI
|
|
20
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okada K, Minamino T and Kitakaze M: Role
of endoplasmic reticulum stress in hypertrophic and failing hearts.
Nihon Yakurigaku Zasshi. 126:385–389. 2005.(In Japanese).
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lai E, Teodoro T and Volchuk A:
Endoplasmic reticulum stress: Signaling the unfolded protein
response. Physiology (Bethesda). 22:193–201. 2007.PubMed/NCBI
|
|
25
|
Yang Y, Zhang Y, Liu X, Zuo J, Wang K, Liu
W and Ge J: Exogenous taurine attenuates mitochondrial oxidative
stress and endoplasmic reticulum stress in rat cardiomyocytes. Acta
Biochim Biophys Sin (Shanghai). 45:359–367. 2013. View Article : Google Scholar : PubMed/NCBI
|