Introduction
Colorectal cancer (CRC), originating from the
epithelial lining of the large bowel (1), is a malignant neoplasm containing colon
and rectal cancer. Its morbidity rate has increased rapidly over
the last 30 years in many countries, including China, Japan and
Spain (2). The treatment of CRC
predominantly includes surgery, chemotherapy, radiation therapy,
biological therapy and the combinational therapy; however, these
treatments are not always entirely effective and so CRC often has a
poor prognosis (3,4). The development of CRC is a multistage
and complex process, as with the majority of malignant tumors. The
mechanisms involved in the development of CRC are not fully
understood and have attracted the interest of many researchers
(5). P16 is a tumor suppressor that
has a key influence in many diseases, including CRC (6). The hypermethylation of P16 has been
demonstrated to have a key role in carcinogenesis (7). The occurrence of CRC is reported to
result from a position named ‘field defect’; however, this
mechanism is not well known (8). It
has been reported that DNA methylation may have a role in mediating
the field defect (8). In CRC cells,
alterations to DNA methylation has an important influence (9). However, the molecular mechanism of how
DNA hypermethylated genes influence the development of cancer has
not been fully elucidated.
To study the varied processes in cancer, such as the
inactivation of the X chromosome and the silencing of tumor-related
genes, it is necessary to understand the details of DNA methylation
in 5′-C-phosphate-G-3′ (CpG) regions (10). CpG islands are often made up of gene
promoters or exons (11). However,
these islands are often unmethylated in normal cells, and
methylation of CpG regions may affect condensed chromatin (12). CpG regions often have roles in
promoting DNA replication origins, which are fundamental to
chromosome organization and duplication (13). They are bimodal and enriched at CpG
islands in mouse and Drosophila. (14). Research has demonstrated that there
are often some molecular abnormalities in tumor tissues and
adjacent tissues, despite them appearing histologically normal
(15). The occurrence of these
abnormalities in the adjacent tissues is named ‘field defect’
(16). Research has indicated that
higher DNA methylation levels of Ras association family member
(RASSF) 1A, adenomatous polyposis coli and human mutL homolog (MLH)
exist in CRC than in adjacent tissues (17).
Three-dimensional (3D) cell culture systems as a
tumor model in vitro is a vital tool in research of DNA
methylation (18). In 3D cultured
pluripotent stem cells, DNA methylation was changed and some genes
were upregulated (19). However, it
is not known whether the culture method affects the DNA methylation
in CpG regions in CRC cells. The present study aimed to identify
the relationship between DNA methylation and CRC, and the influence
of culture method on DNA methylation.
Materials and methods
Cell culture
CRC cell line, DLD-1, was purchased from Renmin
Hospital of Hubei University of Medicine (Shiyan, China). DLD-1
cells were routinely cultivated in RPMI-1640 (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin
at two-dimensional (2D), 3D and orthotopic transplantation (Tis)
stages in a humidified cell incubator (5% CO2) at 37°C.
2D culture refers to DLD-1 culture in RPMI-1640 supplemented with
10% FBS in a common culture dish. 3D culture refers to the culture
of DLD-1 cells in suspension in RPMI-1640 free of FBS in a culture
plate containing Matrigel, which was used to distribute the signal
cells. This means that DLD-1 cells were inoculated into 20 nude
rats (age, 6 months; weight, 100–150 g; Animal Center of Shandong
University, Jinan, China) as previously described (20) to establish a xenograft animal model.
The ratio of male to female rats was 1:1. Rats were housed at an
ambient temperature of 20–22°C with a relative humidity of 50±5%
and a 12 h light/dark cycle. The rats were allowed free access to
food and water until the next procedure was performed. Following
the formation of an orthotopic colorectal tumor, after 14 days,
rats were sacrificed by cervical dislocation under 1% pentobarbital
sodium anesthesia (40 mg/kg; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). Tumor tissue was harvested, cut into small pieces and
rapidly frozen in liquid nitrogen. The present study was approved
by the Institutional Animal Care and Use Committee of Jilin
University (Changchun, China).
Total RNA isolation and methylation
chip genome-wide detection
An RNeasy mini kit (Qiagen, Inc., Valencia, CA, USA)
was used to isolate total RNA from rat tissues, according to the
manufacturer's instructions. The reverse transcription reaction was
carried out with TaqMan MicroRNA Reverse Transcription kit (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) in 15
µl containing 5 µl RNA extract, 0.15 µl 100 mM dNTPs, 1 µl
Multiscribe Reverse Transcriptase (50 U/µl), 1.5 µl 10× reverse
transcription buffer, 0.19 µl RNase inhibitor (20 U/µl), 1 µl of
gene-specific primer and 4.16 µl of nuclease-free water. For
synthesis of cDNA, the reaction mixtures were incubated at 16°C for
30 min, 42°C for 30 min, 85°C for 5 min and stored at 4°C. A total
of 1.33 µl cDNA solution was amplified using 10 µl of TaqMan 2X
Universal polymerase chain reaction (PCR) Master Mix with no
AmpErase UNG (Applied Biosystems; Thermo Fisher Scientific, Inc.),
1 µl gene-specific primers/probe and 7.67 µl of nuclease-free water
in a final volume of 20 µl. Quantitative PCR (TaqMan) analysis was
performed on a TaqMan ABI Prism 7000 Sequence Detection System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The reaction
mixtures were incubated at 95°C for 10 min, followed by 40 cycles
of 95°C for 15 sec and 60°C for 1 min. The relative expression was
normalized to the expression of β-actin (forward,
5′-ACAGAGCCTCGCCTTTGC-3′ and reverse, 5′-GCGGCGATATCATCATCC-3′).
Relative fold changes of gene expression were calculated by the
2−ΔΔCq method (21,22). The
methylation primer was as follows: Forward,
5′-CGTTTTATTTCGGTTTTGTTTTC-3′ and reverse,
5′-CTCGAAATTTAATAAAAACTTCACG-3′. The amplified DNA was cut into
segments by restriction endonuclease (Bethesda Research
Laboratories, Inc., Gaithersburg, MD, USA) and the DNA fragments
were precipitated by precipitation solution PM1 (Promega Corp.,
Madison, WI, USA) and isopropanol (Sigma-Aldrich; Merck KGaA). The
sedimentary DNA was suspended again in precipitation solution PM1
and isopropanol. The resuspended DNA samples were dispersed on
beadchip chips and Human HT-12 v. 4.0 BeadChip (Illumina, Inc., San
Diego, CA, USA) was used for hybridization. Illumina iScan 148
(Illumina, Inc.) was applied to image the BeadChips. Before using
this, Illumina data were reserved on the basis of the Minimum
Information about a Microarray Experiment guidelines in the public
Gene Expression Omnibus database (ncbi.nlm.nih.gov/geo/), accession number GSE
54690.
DNA extraction and quantitative
methylation specific PCR (QMSP)
A Multisource Genomic DNA Miniprep kit (Axygen
Scientific; Thermo Fisher Scientific, Inc.) was applied to extract
total DNA from DLD-1 cells, according to the recommendations of
manufacturer. A CpGenome™ DNA Modification kit (Chemicon; Merck
KGaA) was employed to modify each genomic DNA from samples using
sodium bisulfate according to the manufacturer's protocol. The
methylation levels of MLH, phosphatase and tensin homolog (PTEN),
runt-related transcription factor (RUNX), RASSF, cadherin-1 (CDH1),
O-6-methylguanine-DNA-methyltransferase (MGMT) and P16 were
determined. The regions of β-actin that were short of any CpG
dinucleotide were amplified using a Cyclogene thermal cycler
machine (Techne, Cambridge, UK): 65°C for 5 min, 96°C for 2 min,
65°C for 4 min, 96°C for 1 min, 65°C for 1 min, and 96°C for 30
sec. The primers used were as follows: MLH forward
5′-GGTTGGATATTTYGTATTTTTYGAG-3′ and reverse
5′-AATTACTAAATCTCTTCRTCCCTCC-3′; RUNX forward,
5′-CCTTACGTAGAGGTCACAGTAG-3′ and reverse 5′-CTCCAAGCTGCAAAGTCAC-3′;
CDH1 forward, 5′-AATTTTAGGTTAGAGGGTTATCGCGT-3′ and reverse,
5′-TCCCCAAAACGAAACTAACGAC-3′; MGMT forward,
5′-GGGTTATTTGGTAAATTAAGGTATAGAG-3′ and reverse,
5′-CACCTAAAAATAAAACAAAAACTACCAC-3′; P16 forward,
5′-GAGGGGGTAGGGGGATAT-3′ and reverse,
5′-ACCAATCAACCAAAAACTCCATACTA-3′; β-actin forward,
5′-ACAGAGCCTCGCCTTTGC-3′ and reverse, 5′-GCGGCGATATCATCATCC-3′.
Analysis of differential gene
transcription
Principal component analysis (PCA) may be used to
analyze data tables whose content contains some inter-correlated
quantitative dependent variables. Partek Genomics Suite v. 6.5
(Partek, Inc., St Louis, MO, USA) was used to analyze the gene
expression data. The analyzed data were then corrected and
normalized by quantile normalization and summarization (23). PCA was used for global visualization
of all datasets.
Functional enrichment and pathway
enrichment analysis
MetaCore Bioinformatics software (www.portal.genego.com/) was used to identify the
significant pathways. Data for Annotation, Visualization, and
Integrated Discovery (DAVID; (http://david.niaid.nih.gov),) was used to conduct Gene
Ontology (GO; http://www.geneontology.org/) and Kyoto Encyclopedia
of Genes and Genomes (KEGG; http://www.genome.jp/kegg/ or http://www.kegg.jp/) pathway enrichment analysis.
DAVID is able to combine integrate functional genomic annotations
with intuitive graphical summaries. This contributed to the
explanation on genome-scale datasets by promoting the change from
data collection to biological meaning. GO terms and KEGG pathways
with P<0.01 were selected (24).
The P-values were calculated as follows:
p=(a+ba)(c+dc)(na+c)
Where, n is the number of background genes;
a′ is the gene number of one gene set in the gene lists;
a′ + b is the number of genes in the gene list and
one gene set was also included; a′ + c is the gene
number of one gene list in the background genes; and a′ is
replaced with a = a′-1.
Statistical analysis
Scatter plots of the genome-wide methylation changes
in Tamoxifen-resistant lines and parental was performed. SPSS v.
19.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism 6 (GraphPad
Software, Inc., La Jolla, CA, USA) software were employed to
analyze data. Two sample t-tests were used to differentiate the
mean methylation scores between two samples. A paired t-test was
applied to determine the differences of average sib pair in
methylation scores. P<0.05 was considered to indicate a
statistically significant difference.
Results
Analysis of differential gene
transcription
Partek Genomics Suite v. 6.5 was used to analyze the
gene expression data. The analyzed data were then corrected and
normalized by quantile normalization and summarization. The
datasets were deposited for CRC cells (DLD-1) in 2D, 3D and Tis
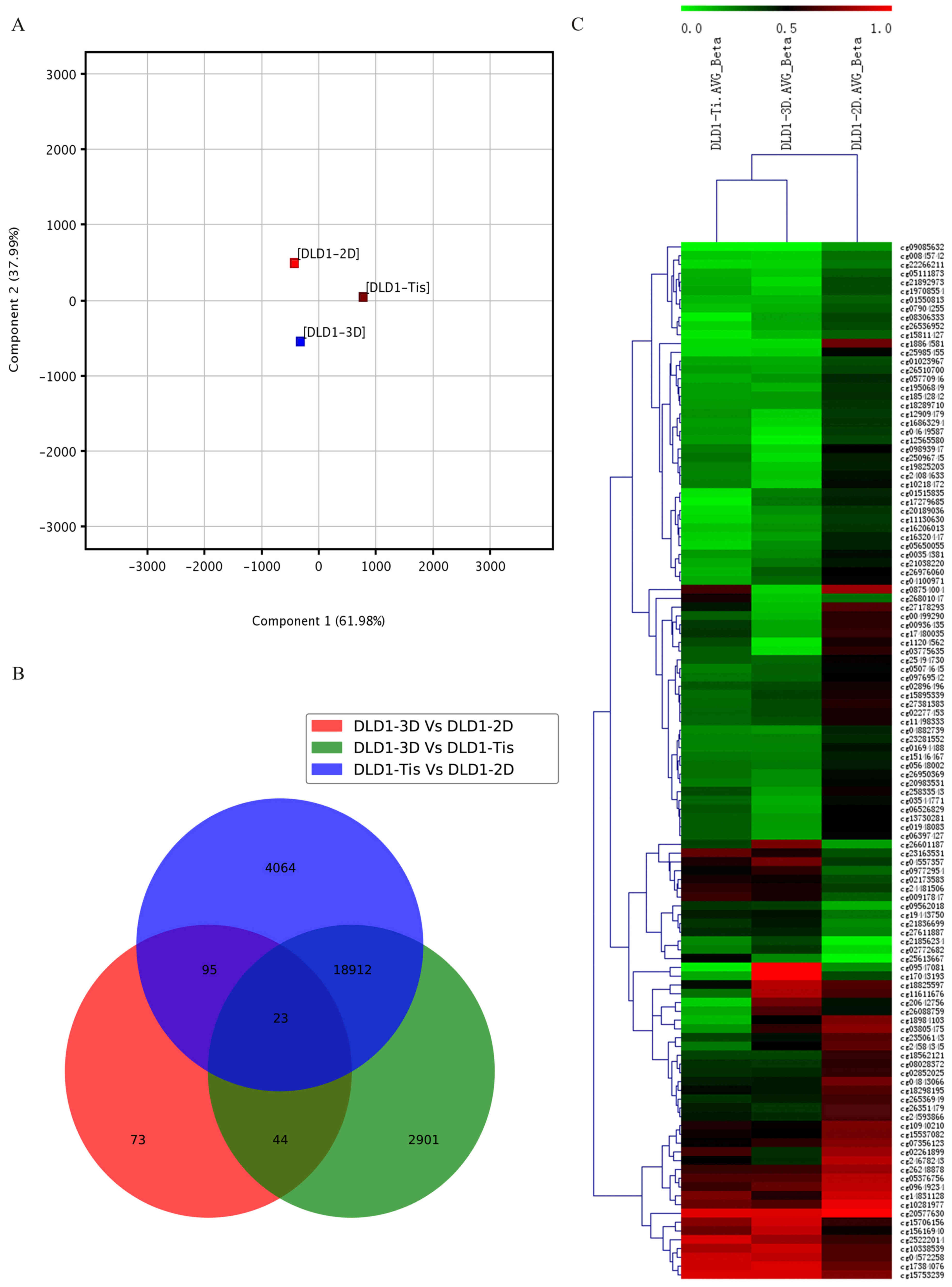
stages. PCA was used for global visualization of all datasets
(Fig. 1A). PCA is one of the most
important and powerful methods in chemometrics as well as in a
wealth of other areas (25). It is a
one-sample technique applied to data with no groupings among the
observations and no partitioning of the variables into sub-vectors
y and × (26). PCA analysis revealed
the close connection between the DLD1-3D, DLD1-2D and DLD1-Tis
groups. Gene lists were established with a cut-off of relative ±
1.5-fold change and P≤0.05. A Venn diagram of modulated samples in
the DLD1-3D, DLD1-2D and DLD1-Tis groups was created. The result
demonstrated that 23 genes were commonly found in the DLD1-3D vs.
DLD1-2D group, DLD1-3D vs. DLD1-Tis group and DLD1-Tis vs. DLD1-2D
group. Apart from the common 23 genes, 95 genes were commonly
identified in the DLD1-3D vs. DLD1-2D group and DLD1-Tis vs.
DLD1-2D group, 44 genes were commonly identified in the DLD1-3D vs.
DLD1-2D group and DLD1-3D vs. DLD1-Tis group, and 18,912 genes were
commonly identified in the DLD1-3D vs DLD1-Tis group and DLD1-Tis
vs. DLD1-2D group (Fig. 1B).
Furthermore, results demonstrated that the overlapping section in
the three populations of samples presented high methylation.
Unsupervised clustering analysis of the CpG location revealed that
119 CpGs presented different levels of methylation in the three
groups of samples and 16 CpGs all appeared with a highly methylated
status in the three groups of samples (Fig. 1C).
Analysis on different CpGs
To analyze the different CpG locations in CRC cells
cultured in 2D, 3D and Tis stage cultures, scatter plots were
created to compare all CpG sites among the DLD1-3D, DLD1-2D and
DLD1-Tis groups (Fig. 2). The areas
outlined in grey on each of the scatter plots in Fig. 2A-C included data points for
demethylated CpG sites that demonstrated a 2-fold change and had
average β values of >0.2. The methylation patterns between
DLD1-3D and DLD1-2D (Fig. 2A) were
more similar than those of DLD1-3D and DLD1-Tis (Fig. 2B), and DLD1-Tis and DLD1-2D (Fig. 2C). Fig.
2D-F demonstrated the column distribution of β values in the
three groups of samples. AVG_β>0.2 stated that the degree of
methylation differences was large.
Functional analysis of differentially
methylated DNA
To identify the biological function, cellular
component and molecular function of the genes identified, GO and
pathway analysis were utilized. From GO, it was demonstrated that
there were differentially expressed genes in various
over-represented cellular processes. In terms of biological
process, DNA-dependent transcription, apoptosis, ion transport,
cell differentiation and transmembrane transport were identified to
be very important. In terms of cellular components, membrane
fraction, cytosol, integral to plasma membrane, perinuclear region
of cytoplasm and cytoskeleton were demonstrated to be important. As
for molecular function, there were some key functions identified,
including signal transducer activity, adenosine 5′-triphosphate
binding, calcium ion binding, protein homedimerization activity and
nucleotide binding (Fig. 3A). To
further refine the biological functions of genes corresponding by
differential methylation sites, these genes were placed into
cellular or metabolic pathways, based on their roles. KEGG may be
used to systematically analyze gene functions based on the networks
of genes and molecules. Pathway analyses of the corresponding genes
recognized 13 significantly over-represented cellular pathways
(P=0.032; Fig. 3B). Among these 13
pathways, four pathways were more significant than the others
(P=0.008). These pathways were cancer pathways, the
mitogen-activated protein kinase (MAPK) signaling pathway, axon
guidance and the insulin signaling pathway.
Culture condition has no effect on
methylation
To determine whether different cell culture methods
affected important genes, a QMSP experiment was used. It has been
reported that PTEN has a role in regulating the formation of
tumors, and its sequence was similar to cytoskeletal protein tensin
(27). RUNX genes have been
identified as tumor suppressors or oncogenes based on their roles
in regulating cell fate and their ambivalent influences in cancer
(28). RASSF genes are tumor
suppressors and RASSF expression is decreased in various types of
cancer (29). The results of the
present QMSP experiment demonstrated that the methylation of the
MLH, PTEN, RUNX, RASSF, CDH1, MGMT and P16 genes and the related
genes had no obvious difference in 2D, 3D and Tis culture
conditions (Fig. 4A and B).
Discussion
CRC has a high incidence and mortality rate
worldwide (30). Studies have
demonstrated that the occurrence rate of CRC in China has increased
yearly and the rate will continue to increase with time (31,32). At
present, ~1.25 million worldwide have CRC and >600,000 patients
will lose their life as a result each year (33). In human CRC, it has been demonstrated
that DNA methylation of CpG islands is able to silence the gene
when the methylation occurs in a promoter region (34). In normal cells, CG nucleotides have
been identified in promoter regions of several tissue-specific
genes with no change in the methylation pattern; however, in cancer
cells, this pattern was altered (35). The present study aimed to explore the
influence of culture method on DNA methylation in CRC.
First, human CRC DLD-1 cells were obtained and
cultured at 2D, 3D and Tis stage cultures. The differentially
methylated DNA in each sample was selected and the relationship
between them was determined by PCA. Results demonstrated that the
differentially methylated DNA in the three different samples was
closely related. In order to determine groups of differentially
methylated sites common in the three samples or belonging to one
sample, a Venn diagram was used for intersection and union analysis
with different comparisons. Results identified that 23
differentially methylated sites were common among the three groups
of samples, and the common sections presented high methylation in
the three samples. Unsupervised clustering analysis was then used
to explore the methylation status of CpG regions. Results
demonstrated that 119 CpGs presented different levels of
methylation in the three groups of samples and 16 CpGs all appeared
to have a high methylation status in the three groups of samples.
Previous research has investigated abnormal DNA methylation in
promoters with a CpG region, and CpG region shores were reported
have a key role in hiding the alteration of DNA methylation in
human CRC (36). In the present
study, scatter plots were created to compare all CpG sites among
the DLD1-3D, DLD1-2D and DLD1-Tis groups and to analyze the
different CpG locations in CRC cells cultured in 2D, 3D and Tis
stage. Results demonstrated that methylation patterns of DLD1-3D
and DLD1-2D were more similar than those of DLD1-3D and DLD1-Tis,
and DLD1-Tis and DLD1-2D.
The results of GO indicated that differentially
expressed genes were involved in molecular function, cellular
component and biological function. KEGG pathway analysis creates
manually curated pathway maps that present content on biological
networks in graphical forms (37).
The present results demonstrated that genes were enriched in 13
pathways, and four of these pathways were more evident than the
rest. These pathways included cancer pathways, the MAPK signaling
pathway, axon guidance and the insulin signaling pathway. PTEN is a
tumor suppressor that may negatively affect the
phosphatidylinositol-3 kinase/protein kinase B signaling pathway
(38). In CRC, abnormal expression
of PTEN is useful for the damage responses to cetuximab (39). Furthermore, PTEN downregulation is
related to liver metastasis and low survival rate in CRC (40). RUNX may regulate a variety of
biological processes, such as growth and differentiation of
lymphocytes and hematopoietic cells (41). In CRC cells, RUNX2 may regulate the
transcription of a metastatic gene, osteopontin (42). RASSF gene family genes have been
reported to be epigenetically silenced with promoter methylation
(43). Ras proteins have an
important role in human cancer and may activate mutations in Ras,
which occur in ~30% of tumors (44).
To explore whether the different cell culture methods had an effect
on important genes, a QMSP experiment was performed in the present
study. Results demonstrated that the methylation of the MLH, PTEN,
RUNX, RASSF, CDH1, MGMT and P16 genes and the related genes had no
obvious difference in the 2D, 3D and Tis culture conditions.
Considering the results and discussion above, in
conclusion, DNA methylation was associated with the development of
CRC; however, this was not altered under 2D, 3D or Tis culture
conditions. However, previous research has demonstrated that DNA
methylation and gene expression in squamous cell carcinoma had
significant differences between 2D and 3D culture systems (45). Compared with squamous cell carcinoma,
CRC may have particular characteristics due to a unique
microenvironment. It is extremely complex to study the molecular
mechanisms of CRC in a tumor model in situ; therefore, 3D
cell culture, may replace animal models as a novel experimental
method to study the progression of CRC. However, the effects of
different culture methods on other cancer types requires further
research.
References
|
1
|
Lobert VH, Mouradov D and Heath JK:
Focusing the spotlight on the zebrafish intestine to illuminate
mechanisms of colorectal cancer. Adv Exp Med Biol. 916:411–437.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Girard P, Ducreux M, Baldeyrou P, Rougier
P, Le Chevalier T, Bougaran J, Lasser P, Gayet B, Ruffié P and
Grunenwald D: Surgery for lung metastases from colorectal cancer:
Analysis of prognostic factors. J Clin Oncol. 14:2047–2053. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Van Cutsem E, Köhne CH, Hitre E, Zaluski
J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G,
et al: Cetuximab and chemotherapy as initial treatment for
metastatic colorectal cancer. N Engl J Med. 360:1408–1417. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang W, Wang PG, Zhan Y and Zhang D:
Prognostic value of p16 promoter hypermethylation in colorectal
cancer: A meta-analysis. Cancer Invest. 32:43–52. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zou HZ, Yu BM, Wang ZW, Sun JY, Cang H,
Gao F, Li DH, Zhao R, Feng GG and Yi J: Detection of aberrant p16
methylation in the serum of colorectal cancer patients. Clin Cancer
Res. 8:188–191. 2002.PubMed/NCBI
|
|
7
|
Merlo A, Herman JG, Mao L, Lee DJ,
Gabrielson E, Burger PC, Baylin SB and Sidransky D: 5′ CpG island
methylation is associated with transcriptional silencing of the
tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med.
1:686–692. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shen L, Kondo Y, Rosner GL, Xiao L,
Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR,
Einspahr JG, et al: MGMT promoter methylation and field defect in
sporadic colorectal cancer. J Natl Cancer Inst. 97:1330–1338. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Müller HM, Oberwalder M, Fiegl H,
Morandell M, Goebel G, Zitt M, Mühlthaler M, Ofner D, Margreiter R
and Widschwendter M: Methylation changes in faecal DNA: A marker
for colorectal cancer screening? Lancet. 363:1283–1285. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Herman JG, Graff JR, Myöhänen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: A novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA. 93:pp.
9821–9826. 1996; View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deaton AM and Bird A: CpG islands and the
regulation of transcription. Genes Dev. 25:1010–1022. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Costello JF, Frühwald MC, Smiraglia DJ,
Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomäki P,
Lang JC, et al: Aberrant CpG-island methylation has non-random and
tumour-type-specific patterns. Nat Genet. 24:132–138. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Holmquist GP and Ashley T: Chromosome
organization and chromatin modification: Influence on genome
function and evolution. Cytogenet Genome Res. 114:96–125. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jones PA and Takai D: The role of DNA
methylation in mammalian epigenetics. Science. 293:1068–1070. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wistuba II, Mao L and Gazdar AF: Smoking
molecular damage in bronchial epithelium. Oncogene. 21:7298–7306.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chai H and Brown RE: Field effect in
cancer-an update. Ann Clin Lab Sci. 39:331–337. 2009.PubMed/NCBI
|
|
17
|
Coppedè F, Migheli F, Lopomo A, Failli A,
Legitimo A, Consolini R, Fontanini G, Sensi E, Servadio A, Seccia
M, et al: Gene promoter methylation in colorectal cancer and
healthy adjacent mucosa specimens: Correlation with physiological
and pathological characteristics, and with biomarkers of one-carbon
metabolism. Epigenetics. 9:621–633. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Godugu C and Singh M: AlgiMatrix™-based 3D
cell culture system as an in vitro tumor model: An important tool
in cancer research. Methods Mol Biol. 1379:117–128. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hiler D, Chen X, Hazen J, Kupriyanov S,
Carroll PA, Qu C, Xu B, Johnson D, Griffiths L, Frase S, et al:
Quantification of retinogenesis in 3D cultures reveals epigenetic
memory and higher efficiency in iPSCs derived from rod
photoreceptors. Cell Stem Cell. 17:101–115. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fidler IJ: The pathogenesis of cancer
metastasis: The ‘seed and soil’ hypothesis revisited. Nat Rev
Cancer. 3:453–459. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bolstad BM, Irizarry RA, Åstrand M and
Speed TP: A comparison of normalization methods for high density
oligonucleotide array data based on variance and bias.
Bioinformatics. 19:185–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ma CH, Lv Q, Cao Y, Wang Q, Zhou XK, Ye BW
and Yi CQ: Genes relevant with osteoarthritis by comparison gene
expression profiles of synovial membrane of osteoarthritis patients
at different stages. Eur Rev Med Pharmacol Sci. 18:431–439.
2014.PubMed/NCBI
|
|
25
|
Bro R and Smilde AK: Principal component
analysis. Anal Met. 6:2812–2831. 2014. View Article : Google Scholar
|
|
26
|
Rencher AC: Principal component analysis.
Met Multi Anal (Second). 1–407. 2002.
|
|
27
|
Stambolic V, Suzuki A, De La Pompa JL,
Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM,
Siderovski DP and Mak TW: Negative regulation of PKB/Akt-dependent
cell survival by the tumor suppressor PTEN. Cell. 95:29–39. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blyth K, Cameron ER and Neil JC: The RUNX
genes: Gain or loss of function in cancer. Nat Rev Cancer.
5:376–387. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Allen NP, Donninger H, Vos MD, Eckfeld K,
Hesson L, Gordon L, Birrer MJ, Latif F and Clark GJ: RASSF6 is a
novel member of the RASSF family of tumor suppressors. Oncogene.
26:6203–6211. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Atkin WS, Edwards R, Kralj-Hans I,
Wooldrage K, Hart AR, Northover JM, Parkin DM, Wardle J, Duffy SW
and Cuzick J: UK Flexible Sigmoidoscopy Trial Investigators:
Once-only flexible sigmoidoscopy screening in prevention of
colorectal cancer: A multicentre randomised controlled trial.
Lancet. 375:1624–1633. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dai Z, Zheng RS, Zou XN, Zhang SW, Zeng
HM, Li N and Chen WQ: Analysis and prediction of colorectal cancer
incidence trend in China. Zhonghua Yu Fang Yi Xue Za Zhi.
46:598–603. 2012.(In Chinese). PubMed/NCBI
|
|
32
|
Ni S, Peng J, Huang D, Xu M, Wang L, Tan
C, SUN H, Cai S and Sheng W: HER2 overexpression and amplification
in patients with colorectal cancer: A large-scale retrospective
study in Chinese population. Am Society Clin Oncol. 2017.
|
|
33
|
Mu WP, Wang J, Niu Q, Shi N and Lian HF:
Clinical significance and association of RUNX3 hypermethylation
frequency with colorectal cancer: A meta-analysis. Onco Targets
Ther. 7:1237–1245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Weisenberger DJ, Siegmund KD, Campan M,
Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D,
Buchanan D, et al: CpG island methylator phenotype underlies
sporadic microsatellite instability and is tightly associated with
BRAF mutation in colorectal cancer. Nat Genet. 38:787–793. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yuan J, Luo RZ, Fujii S, Wang L, Hu W,
Andreeff M, Pan Y, Kadota M, Oshimura M, Sahin AA, et al: Aberrant
methylation and silencing of ARHI, an imprinted tumor suppressor
gene in which the function is lost in breast cancers. Cancer Res.
63:4174–4180. 2003.PubMed/NCBI
|
|
36
|
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z,
Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al:
The human colon cancer methylome shows similar hypo- and
hypermethylation at conserved tissue-specific CpG island shores.
Nat Genet. 41:178–186. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang JD and Wiemann S: KEGGgraph: A graph
approach to KEGG PATHWAY in R and bioconductor. Bioinformatics.
25:1470–1471. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Arico S, Petiot A, Bauvy C, Dubbelhuis PF,
Meijer AJ, Codogno P and Ogier-Denis E: The tumor suppressor PTEN
positively regulates macroautophagy by inhibiting the
phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol
Chem. 276:35243–35246. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Perrone F, Lampis A, Orsenigo M, Di
Bartolomeo M, Gevorgyan A, Losa M, Frattini M, Riva C, Andreola S,
Bajetta E, et al: PI3KCA/PTEN deregulation contributes to impaired
responses to cetuximab in metastatic colorectal cancer patients.
Ann Oncol. 20:84–90. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sawai H, Yasuda A, Ochi N, Ma J, Matsuo Y,
Wakasugi T, Takahashi H, Funahashi H, Sato M and Takeyama H: Loss
of PTEN expression is associated with colorectal cancer liver
metastasis and poor patient survival. BMC Gastroenterol. 8:562008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Miyazono K, Maeda S and Imamura T:
Coordinate regulation of cell growth and differentiation by
TGF-beta superfamily and Runx proteins. Oncogene. 23:4232–4237.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wai PY, Mi Z, Gao C, Guo H, Marroquin C
and Kuo PC: Ets-1 and runx2 regulate transcription of a metastatic
gene, osteopontin, in murine colorectal cancer cells. J Biol Chem.
281:18973–18982. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Djos A, Martinsson T, Kogner P and Carén
H: The RASSF gene family members RASSF5, RASSF6 and RASSF7 show
frequent DNA methylation in neuroblastoma. Mol Cancer. 11:402012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
van der Weyden L and Adams DJ: The
Ras-association domain family (RASSF) members and their role in
human tumourigenesis. Biochim Biophys Acta. 1776:58–85.
2007.PubMed/NCBI
|
|
45
|
DesRochers TM, Shamis Y, Alt-Holland A,
Kudo Y, Takata T, Wang G, Jackson-Grusby L and Garlick JA: The 3D
tissue microenvironment modulates DNA methylation and E-cadherin
expression in squamous cell carcinoma. Epigenetics. 7:34–46. 2012.
View Article : Google Scholar : PubMed/NCBI
|