Introduction
Adriamycin (ADR)-containing chemotherapy is known to
cause dose-dependent and irreversible cardiac damage, which
manifests clinically as a decrease in the left ventricular ejection
fraction (EF) and heart functional deterioration (1). Patients with ADR-induced cardiomyopathy
have a 1-year survival rate of no more than 50% (2). Limiting the use of ADR and
discontinuing the drug are the only clinically accepted methods of
preventing ADR-induced cardiomaopathy (3); thus, treating cancer with ADR is
chanllenging. The pathogenesis of ADR-induced cardiotoxicity is
thought to be driven by the generation of reactive oxygen species
(ROS) (4), as well as fibrosis,
calcium overload and apoptosis (5).
Endoplasmic reticulum stress (ERS) is known to be
involved in the development of many diseases (6,7) and is
characterized by the abnormal accumulation of unfolded and
misfolded proteins (8). ERS can be
activated by external and internal stimuli, such as hypoxia,
oxidative stress, inflammation and toxic compounds (9). In particular, oxidative stress is
believed to be closely related to ERS in the pathogenesis of
numerous diseases. Moderate ERS plays an important role in
maintaining endoplasmic reticulum (ER) function and homeostasis by
enhancing protein folding capacity, while excessive ERS leads to
cell injury and apoptosis (9).
It is well known that adenosine
triphosphate-sensitive potassium channels (K-ATP) are distributed
in various tissues throughout the body, including cardiac muscle,
skeletal muscle, smooth muscle and the brain (10). Channel opening plays a cytoprotective
role under various pathophysiological conditions (11). Glibenclamide (Gli), a K-ATP channel
blocker, has been shown to induce apoptosis and loss of function in
pancreatic β-cell lines (12) by
activating ERS. Gli also impaires the protective effects of
ischaemic pre-conditioning and K-ATP channel opening in the heart.
However, little is known about the role of Gli in ADR-induced
cardiotoxicity. Thus, we sought to investigate the impact of Gli on
ADR-induced cardiotoxicity in rats and the related mechanisms.
Materials and methods
Animals
All procedures involving animals were approved by
the Ethics Committee for Animal Research of Wuhan University. All
animals received humane care in compliance with the Guide for the
Care and Use of Laboratory Animals prepared by the Institute of
Laboratory Animal Resources and the National Research Council. All
animals were acclimated to the laboratory for at least one week
before the experiments.
A total of 60 male Sprague-Dawley (SD) rats (150–180
g) were purchased from the Experimental Animal Center of Wuhan
University and were randomly divided into the following four
groups: i) Control; ii) Gli (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany); iii) ADR (Actavis Italy S.p.A., Nerviano, MI, Italy); and
iv) Gli+ADR (n=15 in each group). The rats in the ADR and Gli+ADR
groups were treated with ADR at a dose of 2.5 mg/kg/week via
intraperitoneal injection for 6 weeks, while the rats in the
control and the Gli groups were treated with normal saline (via
intraperitoneal injection) at the same dose with ADR for 6 weeks.
The rats in the Gli group and the Gli+ADR group received Gli at a
dose of 12 mg/kg/day via gastric lavage for 30 days from the eighth
week of the study, while the rats in the control and the Gli groups
were treated with solvent (via gastric lavage) at the same dose
with Gli for 30 days. The doses of ADR and Gli were selected on the
basis of the previous studies (13,14).
Upon completion of the 30-day Gli or solvent treatment period,
cardiac function was assessed by echocardiography. The rats were
subsequently sacrificed using an overdose of anesthesia (100 mg/kg
pentobarbital), and the cardiac tissues harvested. The cardiac
tissues used for haematoxylin & eosin (H&E) and TUNEL assay
were saved in formalin, and those for western blotting and RT-qPCR
were saved in −80°C. However, the cardiac tissues used for
superoxide dismutase (SOD) and malondialdehyde (MDA) measurements
must be tested as soon as possible after extracted from the
rats.
Echocardiography
Echocardiography was performed using a
high-resolution ultrasound imaging system equipped with a 7V3 probe
with a frequency of 6.0 MHz (Acuson Sequoia 512; Siemens Medical
Solutions, Mountain View, CA, USA). Data pertaining to the
following parameters were recorded: EF%, fractional shortening %
(FS%), left ventricular internal dimension diastolic (LVIDD), left
ventricular internal dimension systole (LVIDS), left ventricular
end diastolic volume (LVEDV) and left ventricular end systolic
volume (LVESV). Data pertaining to the FS%, LVIDD and LVIDS were
recorded from parasternal long-axis M-mode images in accordance
with the American Society of Echocardiography guidelines. The data
represent the average measurements from three to five consecutive
cardiac cycles. The LVEDV and LVESV were calculated from
bi-dimensional long-axis parasternal views by the single-plane
area-length method. The EF% was calculated as follows:
EF%=(LVEDV-LVESV)/LVEDV × 100%.
Histological examination and
TdT-mediated dUTP nick end labeling (TUNEL) assay
Myocardial tissues removed from the middle portion
of each heart were fixed in 10% buffered formalin for 24 h,
embedded in paraffin and sliced into 5 µm-thick sections, which
were then stained with H&E and visualized by light microscopy
for heart size assessments.
The cardiomyocyte apoptosis rate was assessed by the
TUNEL assay. The steps are as below: Sections (3 µm) from
formalin-fixed paraffin-embedded myocardial tissues were
deparaffinized with xylene and dehydrated with ethanol. The slides
were then rinsed twice with PBS and treated with proteinase K (15l
g/ml in 10 mMTris/HCl, pH 7.4–8.0) for 15 min at 37°C. Endogenous
peroxidase activity was blocked with 3% hydrogen peroxide in
methanol for 10 min at room temperature. The tissue sections were
then analyzed with an in situ cell death detection kit (POD;
Roche Diagnostics GmbH, Mannheim, Germany), in accordance with the
manufacturer's instructions. The reactions were visualized with
fluorescence microscopy and measured with a quantitative digital
image analysis system (Image-Pro Plus 6.0; Media Cybernetics, Inc.,
Rockville, MD, USA).
SOD activity and MDA content
measurements
SOD activity in myocardial tissue was detected using
the xanthine oxidase (XO) technique. This procedure depends on the
inhibition of nitrite (NIT) reduction by the superoxide anion,
which is generated by the combination of xanthine and XO. An SOD
assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing,
China) was used to assess SOD activity. One unit of SOD decreased
the rate of NIT reduction by 50%. SOD activity in the myocardial
tissue homogenate was expressed as U/mg protein.
MDA content in myocardial tissue was assayed by the
thiobarbituric acid (TBA) method. This method is based on the
theory that at high temperature (90–100°C) and under acidic
conditions, MDA reacts with TBA to form TBARS, the production of
which was measured at 532 nm by a spectrophotometer. An MDA assay
kit (Nanjing Jiancheng Bioengineering Institute) was used to assess
MDA concentrations. MDA content in the myocardial tissue homogenate
was expressed as nmol/mg protein.
Western blotting and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Cardiac tissues were lysed in RIPA lysis buffer, and
the protein concentration was determined with a BCA protein assay
kit. The protein extracts (30 µg per lane) were separated by
SDS-PAGE and then transferred to polyvinylidene difluoride (PVDF)
membranes, which were probed with various primary antibodies. After
incubating with the appropriate secondary antibodies for 1 h at
room temperature, the membranes were treated with ECL reagents
(Bio-Rad Laboratories, Inc., Hercules, CA, USA), and the signals
were visualized with an Odyssey Imaging System. The expression
levels of specific protein were normalized to those of GAPDH on the
same PVDF membrane. The following primary antibodies were used for
the experiment: Anti-GAPDH antibody; anti-Bax antibody (Epitomics,
Burlingame, CA, USA); anti-Bcl-2 antibody; anti-glucose-regulated
protein 78 (GRP78) antibody; anti-C/EBP homologous protein (CHOP)
antibody, anti-phosphorylated eukaryotic translational initiation
factor 2α (p-eIF2α) antibody, anti-activating transcription factor
6α (ATF6α) antibody and anti-X-box-binding protein 1 (XBP1)
antibody (Cell Signaling Technology, Inc., Danvers, MA, USA).
For RT-qPCR, total RNA was extracted from
ventricular tissues using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), and then first-strand cDNA was
synthesized from the RNA using a Transcriptor First-strand cDNA
Synthesis Kit (Roche Diagnostics, Indianapolis, IN, USA). RT-qPCR
was performed using SYBR-Green PCR Master Mix (Roche Diagnostics)
to determine the expression levels of the genes of interest, and
the results were normalized against the expression levels of GAPDH.
The following primers were used for the experiment: Bax: Forward,
5′-TAGCAAACTGGTGCTCAAGG-3′; and reverse,
5′-TCTTGGATCCAGACAAGCAG-3′. Bcl-2: Forward,
5′-AGCATGCGACCTCTGTTTGA-3′; and reverse,
5′-TCACTTGTGGCCCAGGTATG-3′.
Statistical analysis
All the statistical analyses were performed using
SPSS 18.0 (SPSS, Inc., Chicago, IL, USA). Inter-group comparisons
were analyzed by one-way ANOVA. The data were expressed as the mean
± standard deviation. All P-values were two-sided, and P<0.05
was considered to indicate a statistically significant
difference.
Results
Mortality of rats
Out of 60 rats, 48 completed the study. The
mortality of ADR group and Gli+ADR group were 33.3 and 53.3% at the
end of the interventions, while no deaths were encountered in other
groups.
Gli exacerbates ADR induced
impairments in cardiac function in rats
The H&E staining results indicated that the
increases in heart cross-sectional size induced by ADR were
exacerbated by Gli in the rats in the Gli+ADR group (no data were
obtained) (Fig. 1A).
Echocardiography was performed to measure relative cardiac
functional parameters in each rat. The results consistently
indicated that Gli aggravates ADR-induced impairments in cardiac
function. The LVESV and LVISD in the Gli+ADR group were
significantly larger than those in the ADR group (Fig. 1B and C), while the FS and EF% in the
Gli+ADR group were obviously lower than those in the ADR group
(Fig. 1D and E). However, there were
no significant differences in LVIDD and LVEVD among the four groups
(data not shown).
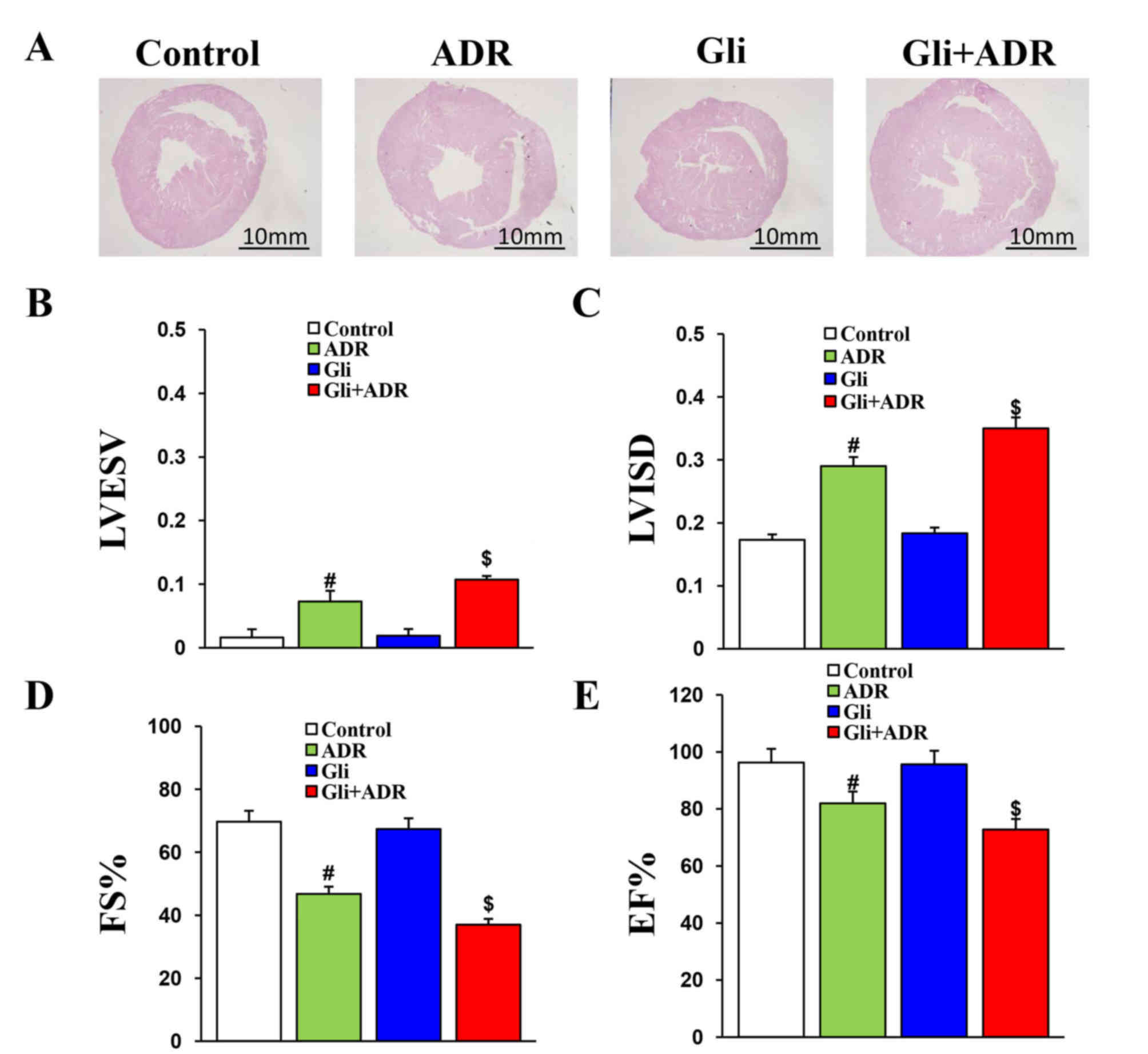 | Figure 1.Gli exacerbates ADR-induced
impairments in cardiac function of rats. (A) Histological analysis
of heart sections from the four groups (scale bar, 20 mm). The (B)
LVESV, (C) LVISD, (D) FS% and (E) EF% data for the four groups.
#P<0.05 vs. Control and Gli groups,
$P<0.05 vs. ADR group. ADR, adriamycin; Gli,
glibenclamide; LVESV, left ventricular end systolic volume; LVISD,
left ventricular internal dimension systole; FS, fractional
shortening; and EF, ejection fraction. |
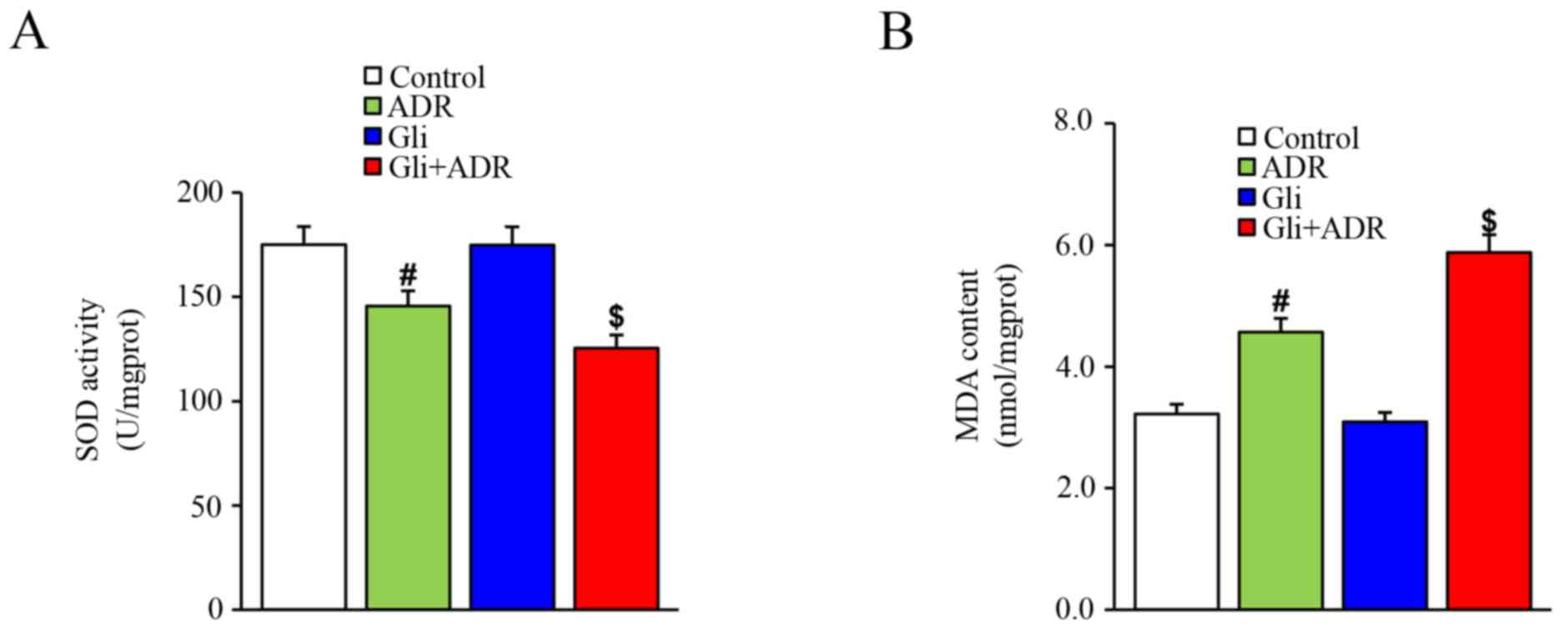
Effect of Gli on oxidative stress in
ADR-treated rats
SOD is the major defense against ROS production in
cells while MDA is the product of the effects of ROS on cell
membrane lipid. Thus, SOD and MDA are used to evaluate oxidative
stress. In this study, ADR elicited a significant decrease in SOD
levels in rat cardiac tissues in the ADR group compared with those
in the control group. Gli treatment decreased SOD levels further in
the Gli+ADR group (Fig. 2A).
However, ADR increased MDA levels in rat cardiac tissues in the ADR
group compared with those in the control group, a change that was
markedly exacerbated by Gli in the Gli+ADR group (Fig. 2B).
The effect of Gli on ERS in
ADR-treated rats
To assess the effect of Gli on ERS, we measured the
expression levels of ERS-related biomarkers by western blotting.
The protein levels of GRP78, CHOP and p-eIF2α in the rats of ADR
group were significantly higher than those in the control group,
but were remarkably lower than those in the Gli+ADR group (Fig. 3A-D). However, there were no
significant differences in ATF6α and XBP1 protein expression levels
among the four groups (Fig. 3A, E and
F). Therefore, Gli exacerbates ADR-induced cardiotoxicity by
activating ERS.
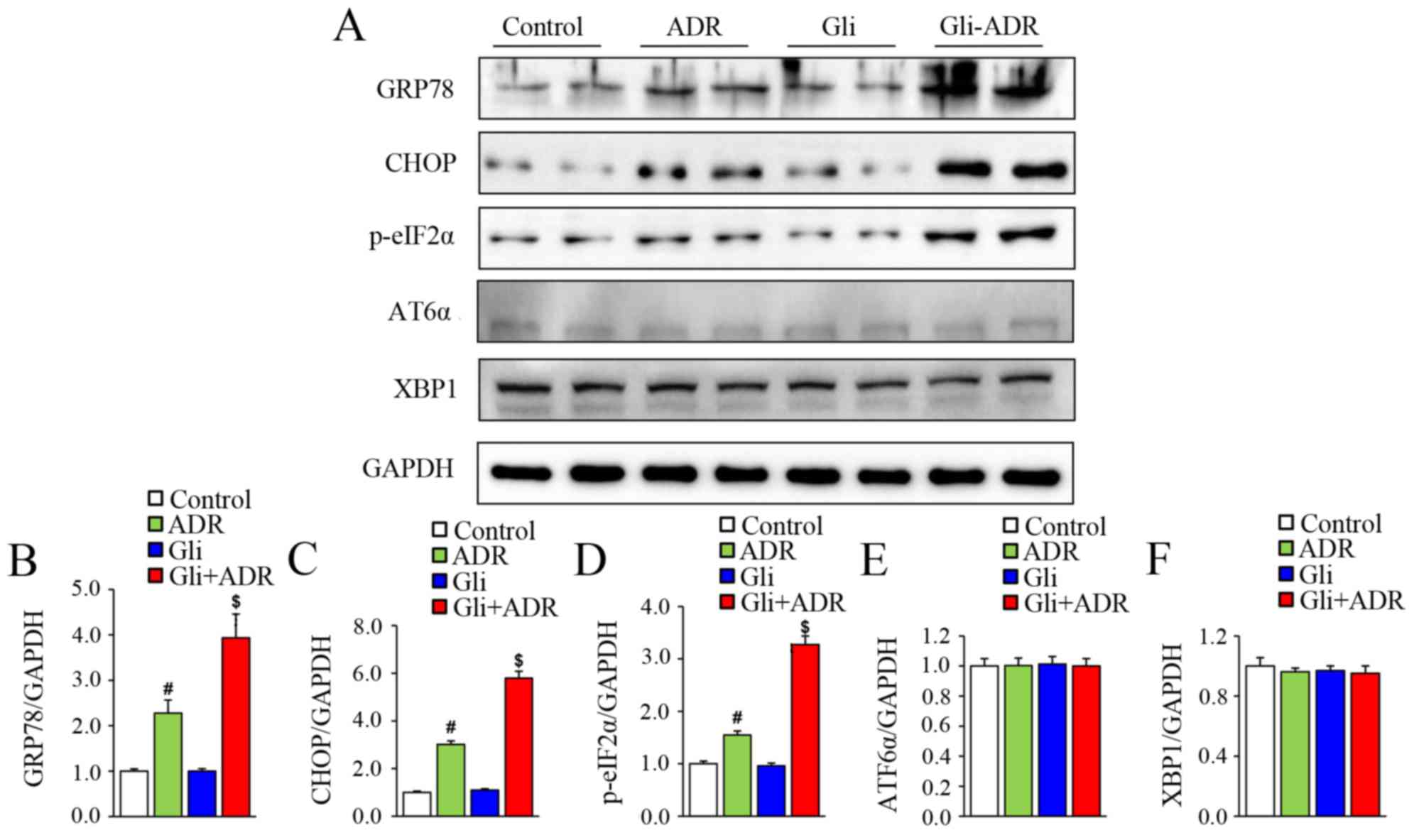 | Figure 3.Effects of Gli on ERS-related
biomarker expression in ADR-treated rats. (A) Western blotting
results for the protein expression levels of GRP78, CHOP, p-eIF2α,
ATF6α and XBP1 in myocardial tissue and the quantified expression
levels of (B) GRP78, (C) CHOP, (D), p-eIF2α, (E) ATF6α and (F)
XBP1. #P<0.05 vs. Control and Gli groups,
$P<0.05 vs. ADR group. ADR, adriamycin; Gli,
glibenclamide; GRP78, glucose-regulated protein 78; p-eIF2α,
phosphorylated eukaryotic translational initiation factor 2α; CHOP,
C/EBP homologous protein; ATF6α, activating transcription actor 6α;
and XBP1, X-box-binding protein-1. |
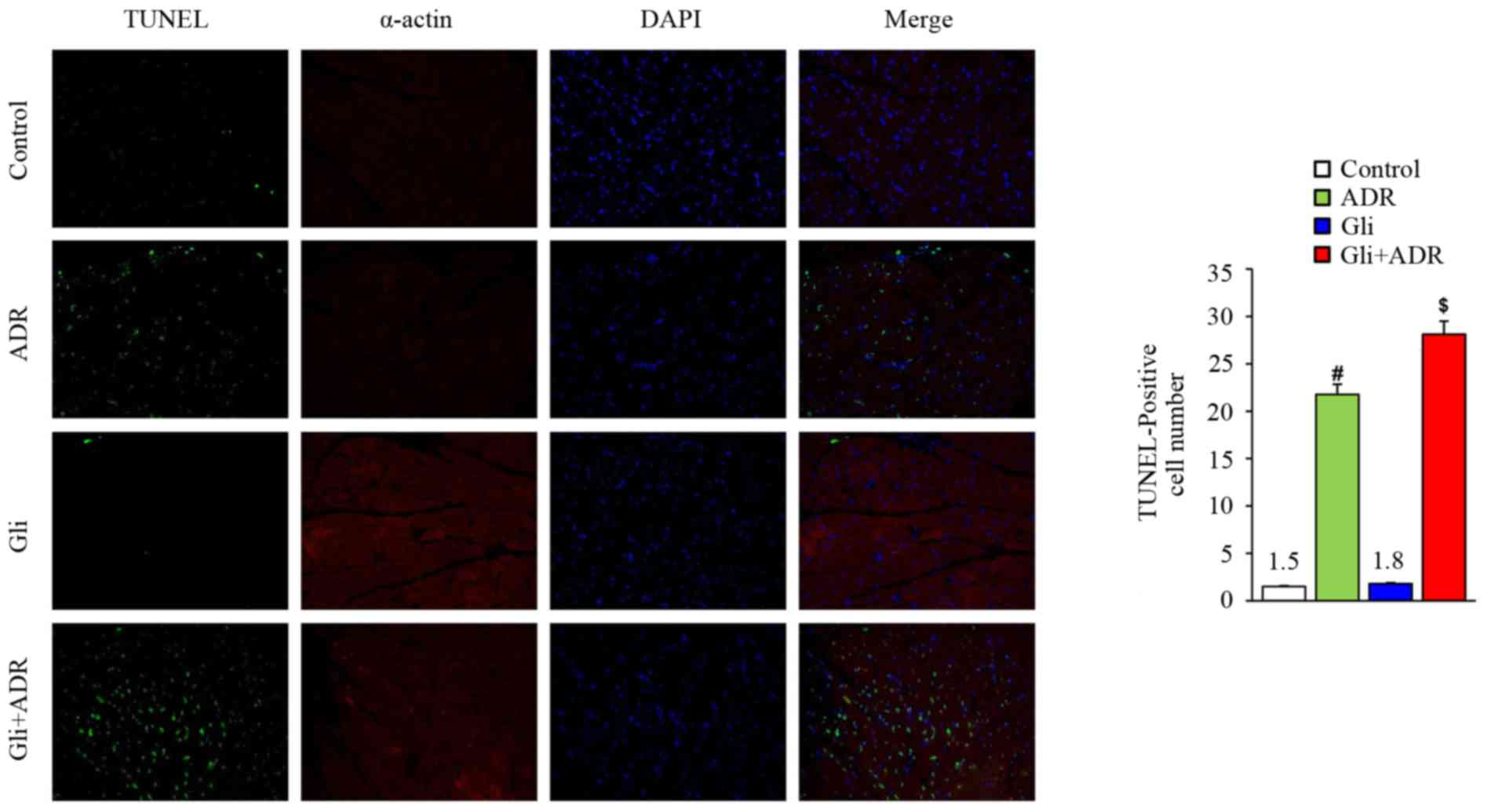
Gli exacerbates ADR-induced myocardial
cell apoptosis in rats
Apoptosis is a type of terminal pathological changes
that occurs in ADR-induced cardiotoxicity. To assess the effect of
Gli on myocardial cell apoptosis, we assessed the expression levels
of apoptosis-related markers. TUNEL staining showed that ADR
increased the cardiomyocyte apoptosis rate in the ADR group
compared with the control group and that Gli increased the
apoptosis rate further in the Gli+ADR group (Fig. 4, scale bar, 50 µm).
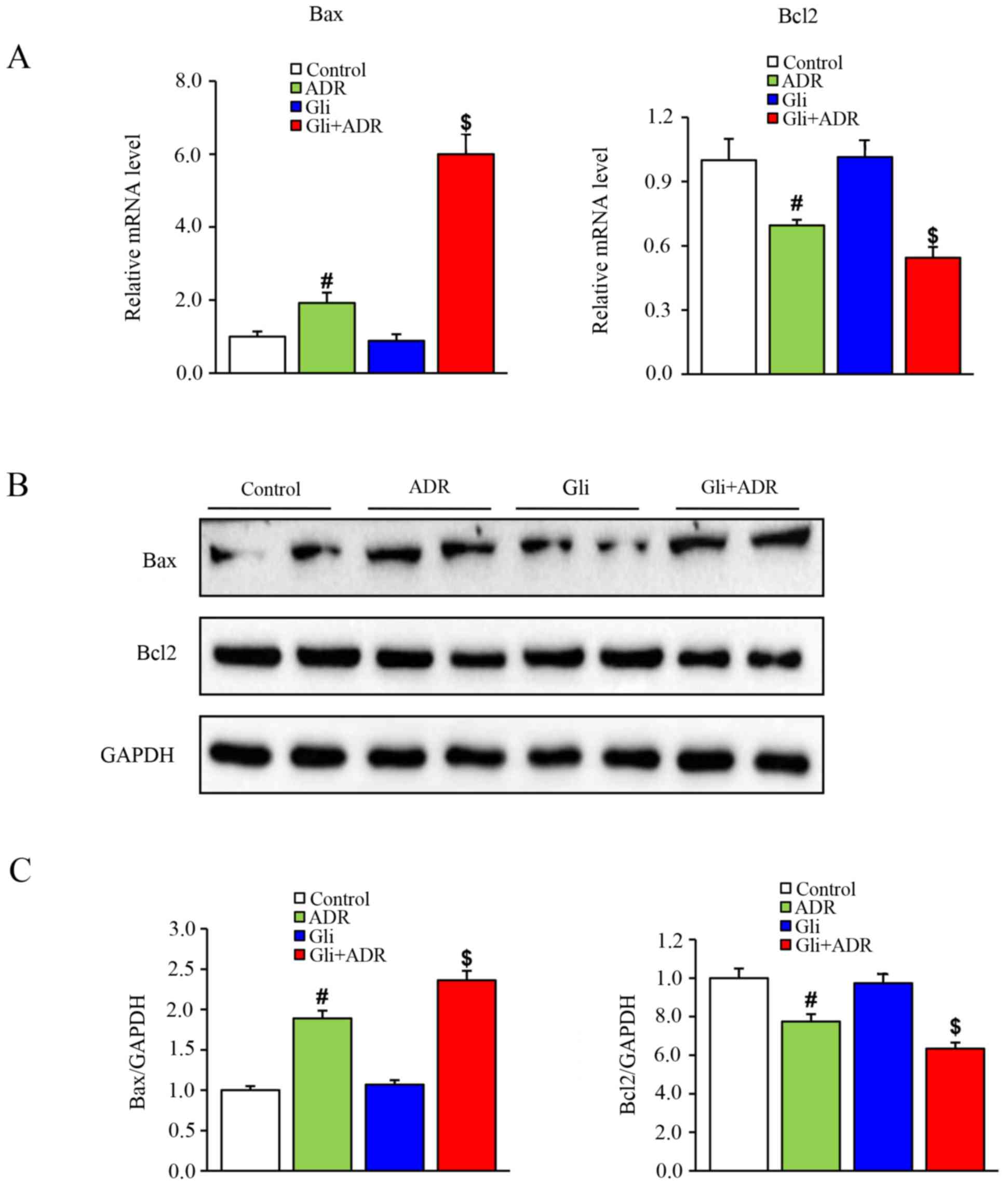
To confirm the above findings, we performed western
blot analysis and RT-qPCR to assess the protein and mRNA expression
levels of apoptosis-related biomarkers, such as Bax and Bcl-2,
respectively. As shown in Fig. 5A,
Bax mRNA expression levels in myocardial tissues in the ADR group
were significantly higher than those in the control group but were
significantly than those in the Gli+ADR group. By contrast, Bcl-2
mRNA expression levels in myocardial tissues in the ADR group were
significantly lower than those in the control group and were
significantly higher than those in the Gli+ADR group.
The western blot analysis results were consistent
with the RT-qPCR results. As shown in Fig. 5B and C, Gli elicited a significant
increase in Bax protein expression levels and a significant
decrease in Bcl-2 protein expression levels in the Gli+ADR group
compared with the ADR and control groups.
Discussion
A previous study showed that Gli (a K-ATP channel
blocker) offsets the cardioprotective effect of nicorandil (a K-ATP
channel opener) in ADR-treated rats (15). In the present study, the highest
mortality was observed in the Gli+ADR group, which was followed by
the ADR group, indicating that Gli could increase mortality induced
by the ADR. Besides, we also observed that the cross-sectional
sizes of rat hearts from the Gli+ADR group were larger than those
from the ADR and control groups (no data were obtained).
Furthermore, more serious heart functional deterioration occurred
in the rats of the Gli+ADR group than in the rats of the ADR group,
as demonstrated by echocardiography. Therefore, our findings were
consistent with those of previous reports.
Oxidative stress is a form of cellular stress and
damage caused by an imbalance between ROS generation and
antioxidant defense mechanisms (16). Oxidative stress generally arises
because of the excessive accumulation of ROS, which overpowers the
antioxidant defences of the body and induces an oxidative reaction
(16). A limited number of ROS are
normally produced by cellular metabolic processes (17). However, excessive accumulation of ROS
or persistent exposure to ROS can lead to the development of many
diseases (18). Previous research
has indicated that oxidative stress is the major mechanism
underlying ADR-induced cardiotoxicity (19). In this study, we observed that ADR
reduced SOD levels and increased MDA levels in rat cardiac tissues
in the ADR group, findings that were consistent with those of the
above mentioned studies. Gli reduced SOD levels and increased MDA
levels further in rat cardiac tissues in the Gli+ADR group. Thus,
we concluded that Gli promotes ROS generation under ADR
stimulation.
The ER is an important cellular organelle in
eukaryotes and participates in the regulation of protein
biosynthesis, folding, transport and modification (20). However, ER-mediated protein folding
is highly and acutely sensitive to intracellular and extracellular
stimuli, such as disruptions of redox homeostasis, ER calcium ions,
changes in energy storage, elevations in mRNA translation and
inflammation (21,22). These phenomena can lead to protein
misfolding. A few misfolded proteins can normally be found in the
ER; however, excessive protein misfolding in the ER can lead to
cellular stress known as ERS (23).
Previous studies have shown that ERS is closely associated with
oxidative stress (18,24) and participates in the pathogenesis of
numerous diseases. Oxidative stress can cause imbalances in
reduction-oxidation (redox) and can activate ERS by decreasing the
efficiency of protein-folding pathways and by promoting protein
misfolding (24). GRP78, an ER
chaperone, is an indicator of ERS and a marker of ERS activation
(9). Under normal conditions, GRP78
is bound to the unfolded protein response (UPR, an appropriate
adaptive response to ERS that relieves ERS by attenuating protein
translation and degrading misfolded or unfolded protein) signal
transducers, such as ATF6, IRE1 and PERK. In the ATF6 pathway,
ATF6α releases from GRP78 and transfers to the nucleus to stimulate
the expression of genes related to the UPR (25). In the IRE1 pathway, IRE1 cleaves and
releases XBP1 mRNA, which is translated into the active XBP1
protein. The activated XBP1 protein then binds to several
UPR-related transcription factors, which leads to target gene
up-regulation (26). In the PERK
pathway, PERK phosphorylates eIF2α and abrogates protein synthesis,
thereby reducing the ER workload to relieve ERS (27). However, phosphorylated-eIF2α
(p-eIF2α) also triggers the synthesis of CHOP (28), which is considered as a vital event
in ERS-induced apoptosis. CHOP has been demonstrated to contribute
to apoptosis induced by cytokines by activating mitochondrial
apoptosis pathways (29). In our
study, we observed that GRP78, CHOP and p-eIF2α protein expression
levels were significantly higher in the rats in the ADR group
compared with control group, but were remarkably lower in the rats
in the ADR group than in those in the Gli+ADR group. Thus, ERS was
activated in rat cardiac tissues damaged by ADR. Gli exacerbates
ERS activation by activating oxidative stress. However, we observed
no significant differences in ATF6α and XBP1 protein expression
levels among the four groups, perhaps because the ATF6 and IRE1
pathways were not activated effectively, and the UPR failed to
relieve ERS.
As a terminal pathophysiological process in cells,
apoptosis has been shown to be induced by a variety of stressors,
such as biomechanical stress, oxidative stress and ERS (30). A previous study reported that ADR
induces myocardial cell apoptosis (31). Consistent with the results of the
above report, our results indicated that the cardiomyocyte
apoptosis rate and Bax protein and mRNA expression levels were
significantly increased, while Bcl-2 protein and mRNA expression
levels were remarkedly decreased in the ADR group compared with the
control group. However, after treated with Gli, the above changes
we detected in the Gli+ADR group were exacerbated further.
Therefore, Gli exacerbates myocardial cell apoptosis in rats
treated with ADR by activating oxidative stress-induced ERS.
Gli, a type of sulfonylurea, has been widely used to
treat type 2 diabetes since the early 1950s by stimulating the
release of insulin from pancreatic β-cells and by reducing blood
glucose levels (32). Our findings
indicated that Gli exacerbates ADR-induced cardiotoxicity by
activating oxidative stress-induced ERS. However, there are
limitations in this study, including that the baseline cardiac
function of rats was not assessed and there wasn't another group
with Gli at another dose. These limitations may affect the
difference during the four groups and let us hard to know the
relationship between the dose of Gil and the effects of Gli on
ADR-induced cardiotoxicity. Despite above limitations, our results
suggest that we should not use sulfonylurea (Gli) and ADR
simultaneously when treating patients with malignant tumours and
type 2 diabetes.
Acknowledgements
This study was supported by grants to Dr. Jun Wan
from the National Natural Science Foundation of China (grant nos.
81170208 and 308711050), and the Natural Science Foundation of
Hubei province, China (grant no. 302-131725).
References
|
1
|
Swain SM, Whaley FS and Ewer MS:
Congestive heart failure in patients treated with doxorubicin: A
retrospective analysis of three trials. Cancer. 97:2869–2879. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takemura G and Fujiwara H:
Doxorubicin-induced cardiomyopathy From the cardiotoxic mechanisms
to management. Prog Cardiovasc Dis. 49:330–352. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jones LW, Haykowsky MJ, Swartz JJ, Douglas
PS and Mackey JR: Early breast cancer therapy and cardiovascular
injury. J Am Coll Cardiol. 50:1435–1441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Octavia Y, Tocchetti CG, Gabrielson KL,
Janssens S, Crijns HJ and Moens AL: Doxorubicin-induced
cardiomyopathy: From molecular mechanisms to therapeutic
strategies. J Mol Cell Cardiol. 52:1213–1225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu
YL, Liu LF and Yeh ET: Identification of the molecular basis of
doxorubicin-induced cardiotoxicity. Nat Med. 18:1639–1642. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Groenendyk J, Sreenivasaiah PK, Kim DH,
Agellon LB and Michalak M: Biology of endoplasmic reticulum stress
in the heart. Circ Res. 107:1185–1197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kwon MJ, Chung HS, Yoon CS, Lee EJ, Kim
TK, Lee SH, Ko KS, Rhee BD, Kim MK and Park JH: Low glibenclamide
concentrations affect endoplasmic reticulum stress in INS-1 cells
under glucotoxic or glucolipotoxic conditions. Korean J Intern Med.
28:339–346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cominacini L, Mozzini C, Garbin U, Pasini
A, Stranieri C, Solani E, Vallerio P, Tinelli IA and Fratta Pasini
A: Endoplasmic reticulum tress and Nrf2 signaling in cardiovascular
diseases. Free Radic Biol Med. 88:233–242. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen Y, Tang Y, Xiang Y, Xie YQ, Huang XH
and Zhang YC: Shengmai injection improved doxorubicin-induced
cardiomyopathy by alleviating myocardial endoplasmic reticulum
stress and caspase-12 dependent apoptosis. Biomed Res Int.
2015:9526712015.PubMed/NCBI
|
|
10
|
Rapposelli S: Novel adenosine
5′-triphosphate-sensitive potassium channel ligands: A patent
overview (2005–2010). Expert Opin Ther Pat. 21:355–379. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Z, Cai H, Dang Y, Qiu C and Wang J:
Adenosine triphosphate-sensitive potassium channels and
cardiomyopathies (Review). Mol Med Rep. 13:1447–1454. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qian L, Zhang S, Xu L and Peng Y:
Endoplasmic reticulum stress in beta cells: Latent mechanism of
secondary sulfonylurea failure in type 2 diabetes? Med Hypotheses.
71:889–891. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schwarz ER, Pollick C, Dow J, Patterson M,
Birnbaum Y and Kloner RA: A small animal model of non-ischemic
cardiomyopathy and its evaluation by transthoracic
echocardiography. Cardiovasc Res. 39:216–223. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Y, Deng QL and Fu ZZ: Effects of
glibenclamide on mRNA level of ATP-sensitive potassium channels of
heart in normal and streptozotocin-induced diabetic rats. Zhonghua
Yi Xue Za Zhi. 80:538–540. 2000.(In Chinese). PubMed/NCBI
|
|
15
|
Abdel-Raheem IT, Taye A and Abouzied MM:
Cardioprotective effects of nicorandil, a mitochondrial potassium
channel opener against doxorubicin-induced cardiotoxicity in rats.
Basic Clin Pharmacol Toxicol. 113:158–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Henriksen EJ, Diamond-Stanic MK and
Marchionne EM: Oxidative stress and the etiology of insulin
resistance and type 2 diabetes. Free Radic Biol Med. 51:993–999.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harding HP, Zhang Y, Zeng H, Novoa I, Lu
PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al: An
integrated stress response regulates amino acid metabolism and
resistance to oxidative stress. Mol Cell. 11:619–633. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stadtman ER, Moskovitz J, Berlett BS and
Levine RL: Cyclic oxidation and reduction of protein methionine
residues is an important antioxidant mechanism. Mol Cell Biochem
234–235. 1–9. 2002.
|
|
19
|
Angsutararux P, Luanpitpong S and
Issaragrisil S: Chemotherapy-induced cardiotoxicity: Overview of
the roles of oxidative stress. Oxid Med Cell Longev.
2015:7956022015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boot-Handford RP and Briggs MD: The
unfolded protein response and its relevance to connective tissue
diseases. Cell Tissue Res. 339:197–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao SS and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress in cell fate decision and
human disease. Antioxid Redox Signal. 21:396–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sharkey LM, Davies SE, Kaser A and
Woodward JM: The role of endoplasmic reticulum stress in intestinal
failure associated liver disease. Clin Nutr ESPEN. 10:e1782015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yuan Y, Xu X, Zhao C, Zhao M, Wang H,
Zhang B, Wang N, Mao H, Zhang A and Xing C: The roles of oxidative
stress, endoplasmic reticulum stress and autophagy in
aldosterone/mineralocorticoid receptor-induced podocyte injury. Lab
Invest. 95:1374–1386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Plaisance V, Brajkovic S, Tenenbaum M,
Favre D, Ezanno H, Bonnefond A, Bonner C, Gmyr V, Kerr-Conte J,
Gauthier BR, et al: Endoplasmic reticulum stress links oxidative
stress to impaired pancreatic Beta-cell function caused by human
oxidized LDL. PLoS One. 11:e01630462016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nadanaka S, Okada T, Yoshida H and Mori K:
Role of disulfide bridges formed in the luminal domain of ATF6 in
sensing endoplasmic reticulum stress. Mol Cell Biol. 27:1027–1043.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Boil. 8:519–529. 2007. View
Article : Google Scholar
|
|
27
|
Kaufman RJ: Regulation of mRNA translation
by protein folding in the endoplasmic reticulum. Trends Biochem
Sci. 29:152–158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Demirtas L, Guclu A, Erdur FM, Akbas EM,
Ozcicek A, Onk D and Turkmen K: Apoptosis, autophagy &
endoplasmic reticulum stress in diabetes mellitus. Indian J Med
Res. 144:515–524. 2016.PubMed/NCBI
|
|
29
|
Allagnat F, Fukaya M, Nogueira TC,
Delaroche D, Welsh N, Marselli L, Marchetti P, Haefliger JA,
Eizirik DL and Cardozo AK: C/EBP homologous protein contributes to
cytokine-induced pro-inflammatory responses and apoptosis in
β-cells. Cell Death Differ. 19:1836–1846. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nugent AE, Speicher DM, Gradisar I,
McBurney DL, Baraga A, Doane KJ and Horton WE Jr: Advanced
osteoarthritis in humans is associated with altered collagen VI
expression and upregulation of ER-stress markers Grp78 and bag-1. J
Histochem Cytochem. 57:923–931. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao S, Li H, Feng XJ, Li M, Liu ZP, Cai Y,
Lu J, Huang XY, Wang JJ, Li Q, et al: α-Enolase plays a
catalytically independent role in doxorubicin-induced cardiomyocyte
apoptosis and mitochondrial dysfunction. J Mol Cell Cardiol.
79:92–103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Del Guerra S, Marselli L, Lupi R, Boggi U,
Mosca F, Benzi L, Del Prato S and Marchetti P: Effects of
pro-longed in vitro exposure to sulphonylureas on the function and
survival of human islets. J Diabetes Complications. 19:60–64. 2005.
View Article : Google Scholar : PubMed/NCBI
|