Introduction
Pheochromocytomas undoubtedly represent an
interesting topic for specialists in fields as diverse as internal
medicine, genetics, histopathology, radiology, urology, and
anaesthesia. Correct and timely diagnosis of pheochromocytomas is
crucial for patients' positive clinical outcome. The management and
treatment of suspected pheochromocytomas usually starts with a
biochemical investigation followed by morphological imaging,
usually computed tomography (CT). In the vast majority of cases,
positive CT findings together with biochemical tests provide
sufficient evidence for the diagnosis of a pheochromocytoma.
The aim of this article is to review recent
multidisciplinary advances in everyday diagnostic and therapeutic
practice, with an emphasis on the role of imaging methods.
Etiopathogenesis
Pheochromocytomas are a rare type of neuroendocrine
tumour originating from the chromaffin cells of the sympathoadrenal
system with permanent or paroxysmal catecholamine hypersecretion
(1,2). The adrenal medulla is the tissue that
is most frequently involved, but the location can also be
extra-adrenal; such tumours are called paragangliomas and are
divided into two groups. Sympathetic paragangliomas, mostly arising
from the abdomen, share their clinical symptomatology with
pheochromocytomas; on the contrary, parasympathetic paragangliomas,
mostly located in the head and neck, could be locally invasive, but
rarely produce catecholamines (1).
The WHO published its latest classification criteria in 2017
(3,4), as well as AJCC publishing the first
staging system for pheochromocytomas and paragangliomas that takes
into account the location of the tumour, size of the primary
tumour, and hormone secretion (5,6). Adrenal
pheochromocytomas constitute 80–85% of cases, with paragangliomas
making up 15–20% of cases in the general population (2,7). Unless
specified otherwise in this review, the term ‘pheochromocytoma’
also refers to sympathetic paragangliomas. The estimated prevalence
of pheochromocytomas is 1:2,500 to 1:1,650, with an annual
incidence between 1,000 and 2,000 cases, including 100–200
pediatric patients and 100–200 with metastatic disease (8).
Genetics
Approximately 40% of pheochromocytomas are of
hereditary origin, the highest degree of heritability amongst any
endocrine tumour type (1,8). Knowledge of the specific genetic
background is nowadays of increasing importance with regard to the
clinical consequences (9–13). Currently, there are at least 12
different genetic syndromes, 15 well-known susceptibility genes,
and an increasing number of potential disease-modifying genes
(8). From the known syndromic
presentations, the most common are syndromes of familial multiple
endocrine neoplasia type 2A (MEN2A) or type 2B (MEN2B),
neurofibromatosis type 1 (NF1), and von Hippel-Lindau syndrome type
2 (VHL 2). Furthermore, familial paraganglioma syndromes are
associated with mutations in genetic encoding for the enzyme
succinate dehydrogenase subunits A, B, C, and D (SDHA, SDHB, SDHC,
and SDHD) (14–17). The association between recurrent,
aggressive, and metastatic paragangliomas and the SDHB gene
mutation is clinically important (18).
Data from the Comprehensive Molecular
Characterization of Pheochromocytoma and Paraganglioma study
proposed a molecular taxonomy of pheochromocytomas and
paragangliomas which has the potential to personalize genetic and
biochemical screening, imaging, follow-up, treatment, and
prevention of the development of a tumour. Patients could be
divided into three main disease clusters: i) pseudohypoxic, ii)
Wnt-signalling, and iii) kinase-signalling (8,19).
The latest Endocrine Society Clinical Practice
Guideline on Pheochromocytoma and Paraganglioma recommends that all
patients with pheochromocytomas should be engaged in shared
decision making for genetic testing. A clinical feature-driven
diagnostic algorithm was created to establish the priorities for
specific genetic testing in pheochromocytoma patients with
suspected germline mutations. Patients with paragangliomas should
undergo testing for SDH mutations and patients with metastatic
disease should undergo testing for SDHB mutations (7).
The number of susceptibility genes is increasing and
their testing with traditional technologies is becoming laborious,
and therefore the application of next-generation sequencing (NGS)
technology might represent the near future for patients with those
tumours. A Consensus Statement with specific recommendations for
the use of diagnostic NGS was published recently (20).
Clinical presentations
Pheochromocytomas and paragangliomas typically
present with symptoms of catecholamine excess. Most of the symptoms
are non-specific, including headaches, palpitations, sweating,
anxiety, nervousness, chest or abdominal pain, nausea, fatigue,
dyspnea, dizziness, intolerance to heat, paresthesia/pain, blurred
vision, constipation, or diarrhoea (9). The typical clinical manifestation of a
pheochromocytoma is sustained or paroxysmal hypertension, and if
the triad of headaches, palpitations, and sweating is accompanied
by hypertension, a pheochromocytoma should be suspected. Other very
common symptoms include orthostatic hypotension, pallor, flushing,
fever, hyperglycemia, vomiting, and convulsions (9). Cardiovascular complications include
hypertensive crises, sudden death, arrhythmias, myocardial
infarction, heart failure resulting from cardiomyopathy (including
tako-tsubo cardiomyopathy), aortic dissection, stroke, non-cardiac
pulmonary oedema, and shock (21).
Unrecognized and untreated pheochromocytomas can lead to
devastating and even fatal consequences (22).
Biochemical tests
The gold standard for the detection of catecholamine
hypersecretion is to measure plasma-free or urinary fractionated
metanephrines (7,22,23).
Free plasma methoxytyramine measurements may be used, if the test
is available, to detect a rare dopamine-producing tumour (24,25).
Therefore optimal screening and follow-up should
include the measurement of metanephrine, normetanephrine,
methoxytyramine, and chromogranin A, to find one of four possible
secretory profiles: Adrenergic, noradrenergic, dopaminergic, and
silent. Catecholamine secretion reflects cell differentiation and
can be used as a prognostic biomarker (8).
If the test results are ambiguous, the clonidine
test may confirm the diagnosis; the test is also able to identify
falsely positive catecholamine elevation (26). It should be noted that the
standardized condition for the blood sampling must be followed and
interfering medication should be avoided.
Imaging methods
The imaging modalities for pheochromocytomas can be
either morphological (US, CT, and MRI) or molecular (scintigraphy
and PET) or a combination of both-the fusion of morphological and
molecular methods (SPECT/CT or PET/CT). Imaging is performed to
determine the location of the tumour following clinical and
biochemical examinations in clinically manifested
pheochromocytomas. A different approach is used in adrenal mass
discovered incidentally by imaging methods; in such a case clinical
and biochemical tests follow imaging.
Morphological imaging
Anatomical imaging methods usually follow clinical
suspicion and positive biochemical tests. Their role is to detect
and locate pheochromocytomas.
The valuable general morphological features that
help identify pheochromocytomas include size, consistency, and
shape. The size may vary from 1 to 15 cm (27), or, rarely, even more. At the time of
the diagnosis, the average size is approximately 4–6 cm (27–32).
Smaller tumours usually consist of solid, relatively homogeneous
tissue (Fig. 1). For larger tumours,
the presence of greater or lesser central necrosis with a
peripheral rim of tumour tissue is typical (Fig. 2). There is also a pure cystic form of
pheochromocytoma.
The shape of a pheochromocytoma is usually spherical
and the edges are relatively smooth.
The diverse morphological appearance of
pheochromocytomas may well mimic other adrenal masses on CT and MRI
scans (33). Differential diagnosis
should aim to distinguish a pheochromocytoma from an adenoma,
metastasis, or adrenal carcinoma.
Ultrasonography
Abdominal sonography may incidentally detect an
adrenal mass, especially on the right side because of the acoustic
window of the liver. Otherwise, the role of ultrasound in the
management of pheochromocytomas is limited.
Computed tomography (CT)
CT is the most common imaging method used in the
diagnosis of pheochromocytomas. Compared to MRI, it is more widely
available, less expensive, and offers better spatial resolution.
The main disadvantage of CT is ionizing radiation. CT scans can
reveal adrenal pheochromocytomas larger than 5–10 mm with
sensitivity >95% (34).
The differentiation of a pheochromocytoma from a
lipid-rich adenoma by means of the unenhanced attenuation value in
Hounsfield units (HU) is straightforward, since the attenuation in
pheochromocytomas is always higher than 10 HU (Fig. 1a) (29). This fact results from an absence of
intra-cytoplasmic lipids within pheochromocytoma tumours, which
also applies to metastases and adrenocortical carcinomas (35). If the average unenhanced attenuation
value of a lesion exceeds 10 HU, it is possible to perform
histogram analysis of the unenhanced CT image; if there are 10% or
more negative pixels in the histogram, an adenoma can be confirmed
as it is thus distinguished from other adrenal lesions, including
pheochromocytomas (36). Currently,
more advanced methods of histogram evaluation (also called ‘CT
texture analysis’) have been introduced (37); according to a recent paper,
pheochromocytomas had a significantly higher mean grey-level
intensity, entropy, and mean of positive pixels, but lower skewness
and kurtosis in unenhanced images compared to lipid-poor adenomas
(32).
After the administration of an iodine contrast
medium, pheochromocytomas usually display pronounced enhancement,
often more than 130 HU. In smaller solid lesions, the enhancement
is relatively homogeneous (Fig. 1b),
while in larger lesions the character of the enhancement is more or
less heterogeneous; something that is very typical of
pheochromocytomas with central necrosis is the pronounced
enhancement of the peripheral rim of the viable tumour tissue
(Fig. 2). Although strong
enhancement occurs in most pheochromocytomas, it cannot be
considered specific, since there is significant overlap of contrast
enhancement with other types of adrenal lesions (29); only a single article has reported
significant differences in the enhancement of adenomas and
pheochromocytomas (38).
The targeted adrenal CT protocol usually includes
late-enhancement scans (i.e., 7–15 min after the application of a
contrast medium), which allows the measurement of the absolute and
relative decreases in post-contrast attenuation, which reflect the
rate of contrast medium washout. Adenomas are believed to express
rapid washout compared to the slower washout of pheochromocytomas
(39–42). However, according to some papers, up
to one third of pheochromocytomas overlap with adenomas in terms of
their relative or absolute washout rate (Fig. 1a-c) (30,31).
Furthermore, washout rates are unable to distinguish
pheochromocytomas from adrenal carcinomas or metastases (41).
Studies with a non-ionic contrast medium (Iohexol)
failed to confirm the widespread opinion that the intravenous
administration of an iodinated contrast medium can lead to
increased secretion of catecholamines from pheochromocytomas or may
even lead to a hypertensive crisis (43). This result was replicated by another
study providing evidence that the administration of iodinated
non-ionic contrast media in patients with a suspected or known
pheochromocytoma is safe, and the administration of α-adrenergic
receptor blockers before the administration of the contrast medium
is not necessary (44).
Magnetic resonance imaging
MRI is not a first-choice imaging tool because of
its lower spatial resolution, lower logistical availability, higher
price, and stricter safety regulations. But it benefits from being
free of ionizing radiation and therefore suitable e.g., in cases of
pregnant women or children or in patients with adverse reactions to
an iodinated contrast medium.
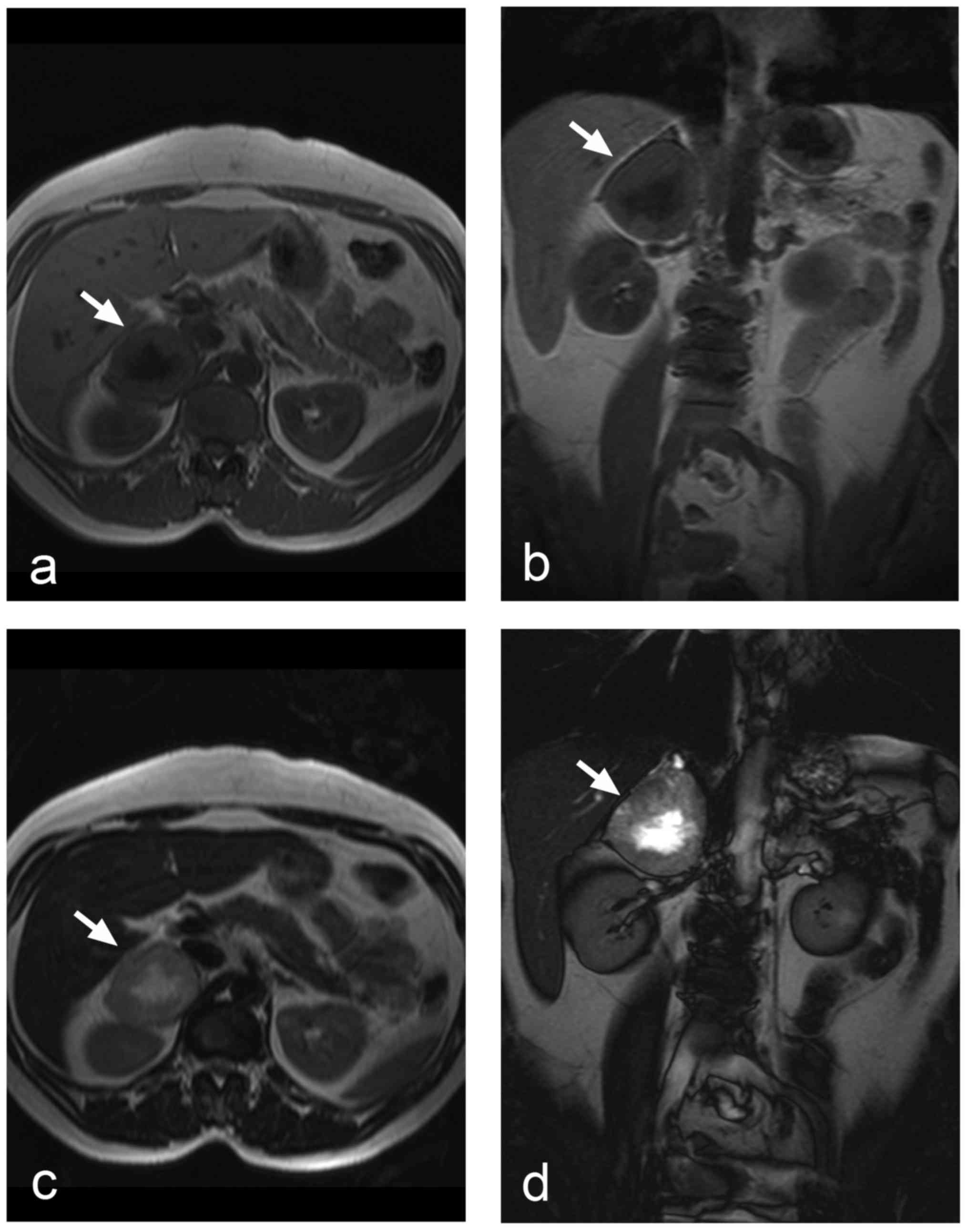
The appearance of pheochromocytomas in
T1- and T2-weighted images depends on whether
the tumour is solid, cystic, or haemorrhagic/necrotic. Cystic
tumours display high signal intensity in T2-weighted
images. A similar picture is seen in pheochromocytomas with central
necrosis (Fig. 3), but the classical
pattern of a T2 hyperintense pheochromocytoma is relatively
uncommon (27). The signal intensity
of the haemorrhage in T1- and T2-weighted
images could vary considerably, because the signal changes over the
time since it depends on the age of the haematoma. However,
generally speaking, the signal intensity of blood is predominantly
high in T1-weighted images. Smaller solid
pheochromocytomas could be distinguished from adrenal adenomas by
means of chemical shift imaging, because unlike adenomas,
pheochromocytomas contain no intracellular lipids, and thus they
show no signal changes in out-of-phase and in-phase images
(Fig. 4a and b) (45).
Another option of MRI is diffusion-weighted imaging
(DWI) and calculation of apparent diffusion coefficient (ADC) maps
(Fig. 4c and d). Significantly
higher ACD values were observed in pheochromocytomas compared to
adenomas and metastases (46). ADC
histogram analysis was also applied and revealed significant
differences between pheochromocytomas and adenomas (47). ADC values might also help to
distinguish benign from malignant pheochromocytomas (48).
MR spectroscopy of pheochromocytomas has been
studied but is not routinely used (49,50).
Although the use of paramagnetic contrast agents is
rarely necessary, strong post-contrast enhancement of
pheochromocytomas is similar to contrast-enhanced CT (51).
Paramagnetic contrast agents used in MRI do not lead
to the hypersecretion of catecholamines (52).
Molecular imaging in the detection of
pheochromocytomas
After the morphological imaging studies have been
obtained, the functional imaging modality could be utilized to
confirm the source of the increased production of catecholamines.
For most of the cases one of the following methods is
employed-123I-MIBG scintigraphy, 18F-FDG, or
18F-DOPA PET/CT and somatostatin receptor imaging. The
selection of the functional modality could be based on knowledge of
the patient's genetic background, as recommended by the European
Association of Nuclear Medicine (53).
123I-MIBG scintigraphy
For a long time, radioactive iodine-labelled
123I-metaiodobenzylguanidine (123I-MIBG) has
been used to reveal pheochromocytomas. MIBG is a guanethidine
precursor and its structure resembles that of norepinephrine.
Following intravenous application, it is transported by the
reuptake mechanism into presynaptic adrenergic neuron cells, where
it accumulates in catecholamine secretory granules through the
adenosine triphosphate system (ATPase-dependent proton pump). The
scintigraphic examination uses the intravenous application of
123I-MIBG tracer. The sensitivity of
123I-MIBG ranges between 85 and 88% for
pheochromocytomas and between 56 and 75% for paragangliomas,
whereas its specificity ranges from 70 to 100% and 84 to 100%,
respectively. Its sensitivity for metastatic pheochromocytomas is
between 56 and 83%, whereas for recurrent disease it is ~75%
(7).
Physiological MIBG uptake occurs in the salivary
glands, the heart, the liver, and the spleen. Slightly elevated
accumulation is also seen in the thyroid gland. Because of the
renal excretion of the tracers, radioactivity is observed in the
kidneys and the urinary bladder. The varying level of accumulation
can also be observed in the nasal mucosa, neck muscles, lungs, and
intestine. Medication which can prevent MIBG from accumulating in
tumours (e.g., insulin, reserpine, amphetamine, calcium channel
blockers, sympathomimetics) should be discontinued before the
examination (54).
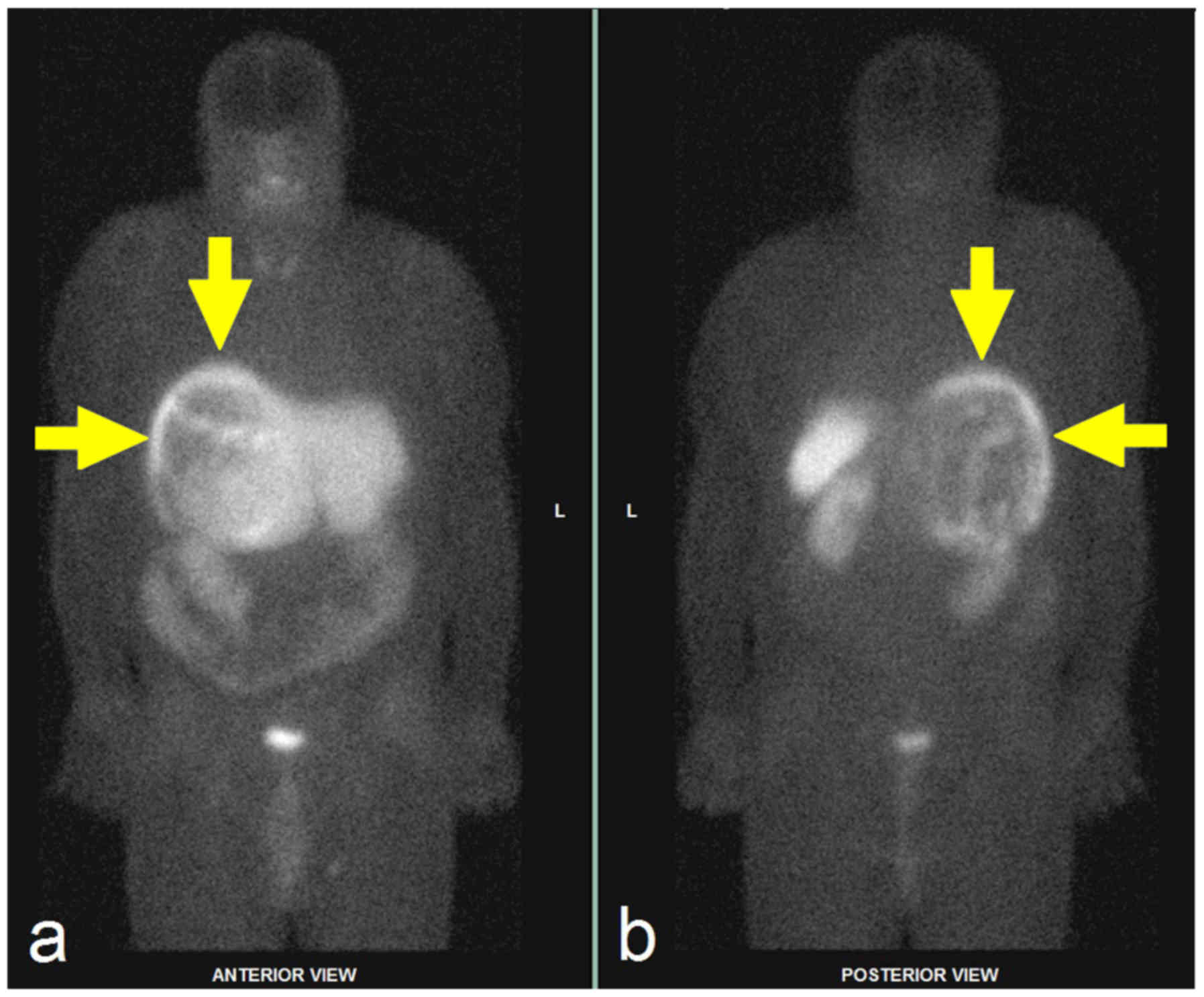
Pheochromocytomas appear on scintigrams as focal
increased concentrations of radioactivity in the adrenal medulla
but also in ectopic adrenergic tissue or metastases (Fig. 5). Paragangliomas can easily be missed
on CT and MRI scans.
The advantages of 123I-MIBG are
high-quality examination with less exposure to radiation and,
compared to the later methods, its wide availability and relatively
low cost.
Nowadays the 123I-MIBG examination is
mostly recommended for patients with metastatic paragangliomas
detected by other imaging modalities, when radiotherapy using
131I-MIBG is planned. Occasionally, it is used in some
patients with an increased risk of metastatic disease because of
the large size of the primary tumour or extra-adrenal, multifocal
(except paragangliomas of the skull base and neck), or recurrent
disease (7).
Somatostatin receptor
scintigraphy
Another option in the visualization of
pheochromocytomas is the detection of somatostatin receptors, whose
concentration is increased in neuroendocrine tumours, including
pheochromocytomas (Fig. 6). This
imaging method uses radionuclide-labelled peptides, specifically
somatostatin analogues (111In-pentetreotide,
99mTc-HYNIC-TOC). Somatostatin receptor imaging is
mainly used in paragangliomas because of the relatively high
physiological uptake of the radiopharmaceutical in the kidneys.
This method is not recommended for hereditary tumours.
PET/CT
In most published examination schemes PET/CT with
corresponding tracers is the preferred method for the detection of
pheochromocytomas and paragangliomas and further wide use of this
method can be expected. The most commonly used radiopharmaceuticals
in this PET scanning are 18F-3, 4-dihydroxyphenylalanine
(18F-DOPA) and 18F-fluorodeoxyglucose
(18F-FDG); the selection of the tracer is based on
localization of the tumour and its genetic background (7,53).
The advantage of 18F-DOPA imaging is the
lack of significant uptake in the normal adrenal medulla. The
efficiency of 18F-DOPA imaging of paragangliomas depends
on the localization of the tumour and genetic status.
18F-DOPA PET/CT is an excellent diagnostic tool for head
and neck paragangliomas, but its sensitivity can be lower in
retroperitoneal paragangliomas (55). In metastatic disease the
18F-DOPA PET detection rate of the lesions is higher in
SDHB-negative patients than in SDHB-positive ones (56).
The visualization of paragangliomas using
18F-FDG PET/CT is influenced by the degree of tissue
differentiation, localization of the tumour, and genetic status
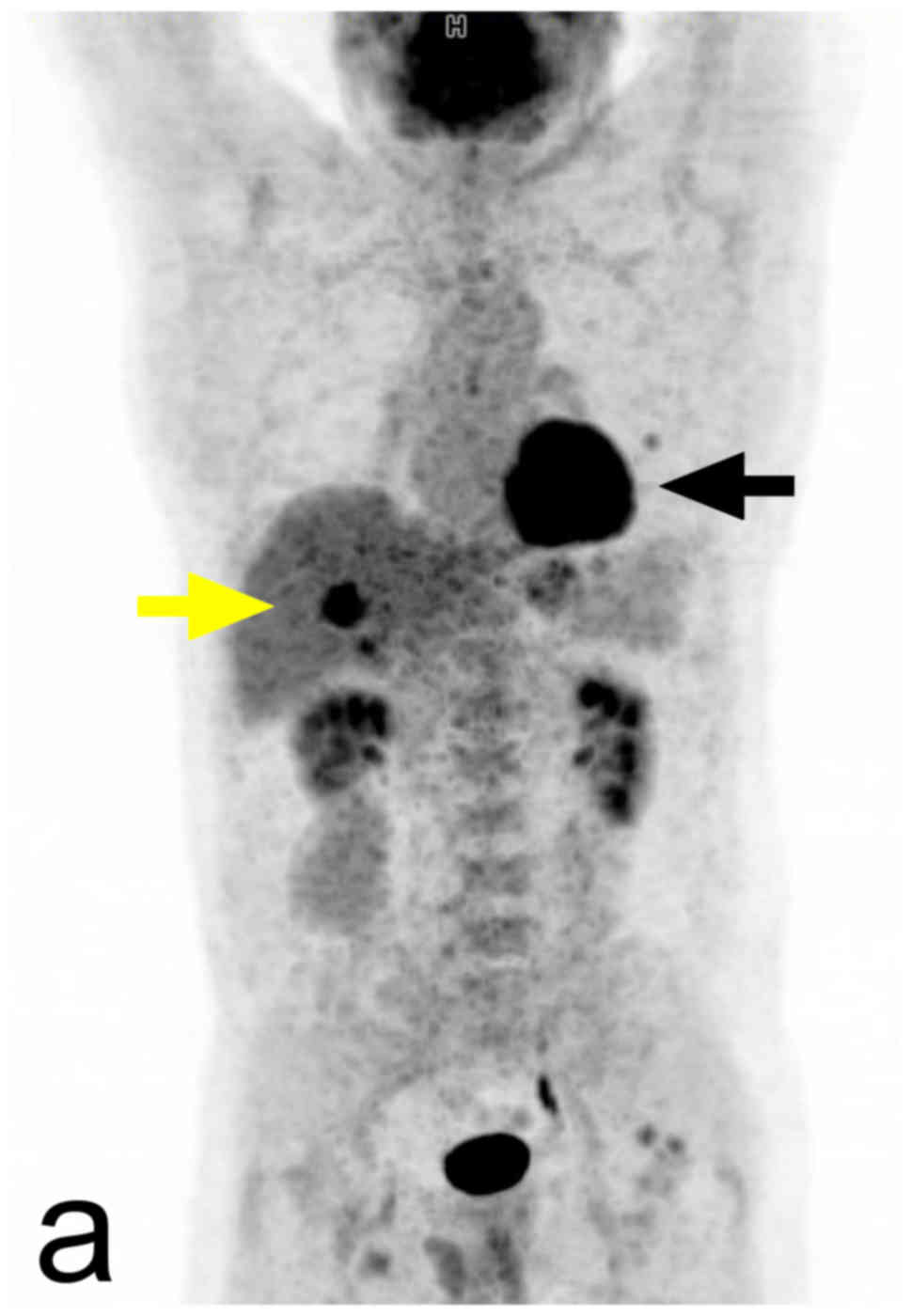
(53). Increased 18F-FDG
uptake is a usual finding in pheochromocytomas, but the intensity
of this uptake is variable. The sensitivity of 18FDG
PET/CT in the detection of pheochromocytomas is high, but
unfortunately, its specificity is lower. In metastatic disease the
18F-FDG PET/CT detection rate of the lesions is higher
in SDHB-positive patients (Fig. 7)
(contrary to 18F-DOPA PET) (57). In patients with known metastatic
pheochromocytomas, 18FDG PET/CT is preferred over
123I-MIBG. 123I-MIBG is performed in this
group of patients when treatment with 131I-MIBG is being
considered (7).
18F-fluorodopamine (18F-FDA)
and 68Ga-DOTA-peptides that specifically bind to
somatostatin receptors are experimental PET tracers that have been
successfully used in clinical studies (39). 68Ga-DOTA-peptides seem to
represent the near future of the molecular imaging of
pheochromocytomas. Their superiority in the localization of
sporadic metastatic pheochromocytomas compared to all other
functional and anatomical imaging modalities has been demonstrated
(58–60).
Molecular imaging-tailoring of the
diagnostic strategy according to guidelines (7,53)
Sporadic pheochromocytomas-the sensitivity of
123I-MIBG is similar to the case of PET imaging and
superior to somatostatin imaging.
Head and neck paragangliomas-the most sensitive
method is 18F-DOPA PET/CT, but according to new studies
imaging using 68Ga labelled somatostatin analogues
achieves similar results. The usage of 18FDG PET/CT is
beneficial in SDHx-related head and neck paragangliomas.
Retroperitoneal paragangliomas-the sensitivity of
18F-DOPA PET/CT is higher than 123I-MIBG
scintigraphy, and 18F-FDOPA PET/CT is more specific than
18FDG PET/CT. High sensitivity is exhibited by
18FDG PET/CT in SDHx- and VHL-related sympathetic
paragangliomas.
Metastatic pheochromocytomas and
paragangliomas-18F-FDG PET/CT is the preferred method in
SDHB-related patients and the usage of 18F-FDOPA PET/CT
can be advantageous in the absence of SDHB mutations; in patients
with unknown genetic status 18F-FDG PET/CT should be
used, optionally in combination with 18F-FDOPA PET/CT or
somatostatin receptor imaging. 123I-MIBG allows the
appropriateness of radiotherapy to be assessed using
131I-MIBG.
Histopathology
An important aspect of the clinical management of
pheochromocytomas is the distinction between benign and malignant
variants. In most cases, it is impossible to predict the biological
properties of pheochromocytomas by imaging methods alone. Malignant
variants can be reliably identified with imaging methods alone only
in the presence of distant metastases.
Pheochromocytomas are tumours of the
sympathochromaffin system. The histological diagnosis is
straightforward thanks to the characteristic morphological pattern.
What is not easy, however, is the prediction of their biological
behaviour on the basis of histological criteria alone.
Histological signs of potential malignancy include a
diffuse infiltrative growth, vascular and capsular invasion, tumour
necrosis, increased mitotic activity, cellular polymorphism, and
high proliferative activity as shown by immunohistochemistry. The
histological evaluation must be comprehensive and must include
immunohistochemical assessment of proliferative activity. If Ki67
>3% it is considered a useful parameter for predicting malignant
potential (61).
Pheochromocytoma of the Adrenal gland Scaled Score
(PASS) <4 had benign behaviour and all malignant cases had PASS
≥6. A PASS score between 4 and 6 needs long-term follow-up. Some
recent studies state that the values are <4 for benign tumours
and ≥6 for malignant tumours, whereas a value between 4 and 6
suggests an intermediate risk (62).
A study performed on 100 cases stated that PASS can be used to
separate tumours with the potential for biologically aggressive
behaviour (PASS≥4) from those that behave in a benign fashion (PASS
<4) (63).
In another study conducted on 11 patients, it was
found that a PASS score ≥4 identifies malignant pheochromocytomas
with a sensitivity of 50% and specificity of 45%. On the basis of
this study it was suggested that PASS helps to reserve the more
aggressive treatment and narrow the follow-up for potentially
malignant tumours (64). It is known
that the size and weight of the pheochromocytomas are directly
related to PASS and malignancy (65).
Malignant pheochromocytomas and adrenocortical
carcinomas can easily be mistaken for one another (66), which often leads to clinically
significant consequences for the patient. Therefore, long periods
of follow-up are necessary for patients following the surgical
excision of pheochromocytomas (67).
The average five-year survival rate for malignant pheochromocytomas
ranges from 34 to 60% (9).
Therapy
Surgical removal of the tumour is currently the only
treatment.
Preoperative management
The risk of catecholamine hypersecretion is
significantly increased by biopsy. Therefore, needle biopsy should
be avoided in clinically suspected pheochromocytomaπs and surgery
should be performed directly (68,69).
Preoperative pharmacological management of the
pheochromocytoma is always needed to reduce the risk of
perioperative complications. Pharmacological preparation of the
patient significantly reduces the risk of perioperative mortality
(70,71). Crucially, the patient should have an
alpha-blocker administered for at least 14 days prior to surgery to
reduce the risk of vasoconstriction. A beta-blocker can be
administered prior to surgery as well to prevent tachycardia, if
necessary. The administration of beta-blockers must not begin prior
to alpha-blockade because of the risk of severe hypertension. An
experienced anaesthesiologist should be present during the surgery
to reduce the risk of possible circulatory complications.
Surgical technique
The surgical method of choice in the treatment of
pheochromocytomas is laparoscopic adrenalectomy, performed via the
trans- or retroperitoneal approach. The retroperitoneal approach is
particularly suitable for the right adrenalectomy.
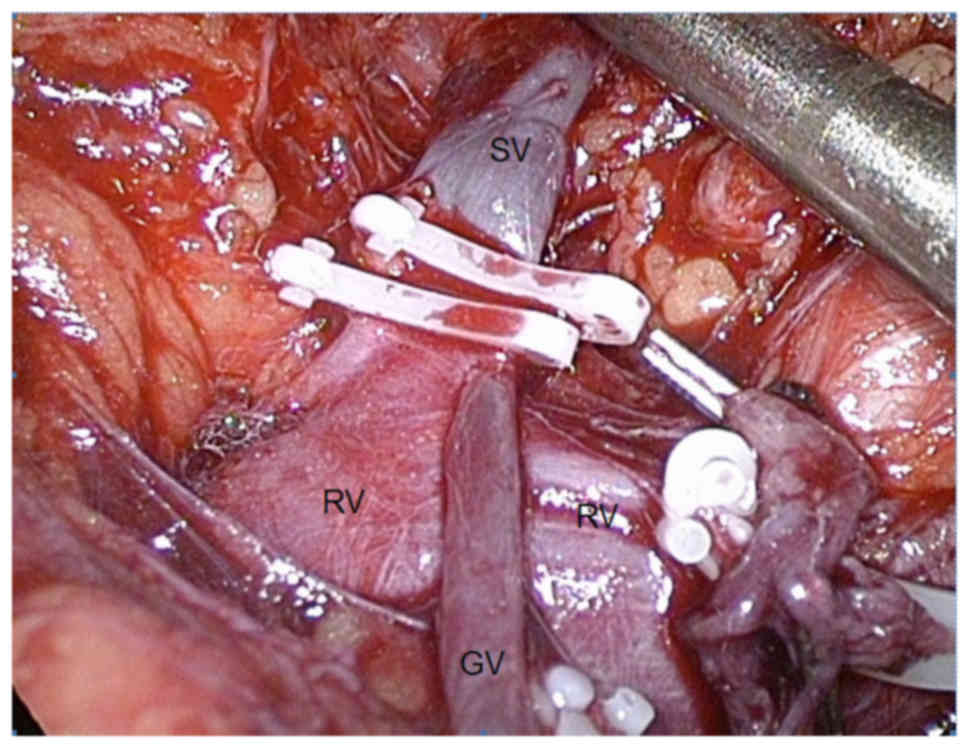
The rule of thumb for preventing complications
during the resection of pheochromocytomas is to perform an early
occlusion of the vein draining the adrenal medulla (known as the
vena centralis), flowing into the renal vein on the left side
(Fig. 8) and into the inferior vena
cava on the right side. This is to minimize the secretion of
catecholamines into the circulation during manipulation of the
gland. Some well-established advantages of the laparoscopic
approach include lower blood loss, less need for narcotics, a
shorter hospital stay, and a more favourable cosmetic effect. The
previous assumption that the intra-abdominal insufflation of carbon
dioxide may induce hypertension has been refuted.
Open adrenalectomy is currently indicated only in
tumours with signs of invasion into the surrounding structures or
in the presence of a thrombus. The open approach is also preferred
in tumours larger than 8–12 cm (72)
and in the event of paraganglioma (7). In cases of a bilateral
pheochromocytoma, e.g., in patients with VHL syndrome or MEN2A,
partial adrenalectomy can be performed at least on one side to
preserve the secretion of adrenal hormones.
In the long-term prognosis of patients after surgery
is excellent; however, hypertension persists in almost 50% of cases
(9).
Conclusion
Pheochromocytomas present important and interesting
clinical features. Their management requires the cooperation and
long-standing experience of several specialists. Timely and correct
diagnosis of this condition is essential for the positive clinical
outcomes of patients. In most cases, correct interpretation of the
abdominal CT scan together with positive biochemical findings
provides sufficient evidence for diagnosis and helps guide
subsequent therapeutic decisions.
Acknowledgments
The present study was supported by the Ministry of
Health of the Czech Republic (grant no. 17-31847A) and the Ministry
of Health, Czech Republic-conceptual development of research
organization (FNOL, 00098892).
References
|
1
|
Dahia PL: Pheochromocytoma and
paraganglioma pathogenesis: Learning from genetic heterogeneity.
Nat Rev Cancer. 14:108–119. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lenders JW, Eisenhofer G, Mannelli M and
Pacak K: Phaeochromocytoma. Lancet. 366:665–675. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lam AK: Update on adrenal tumours in 2017
world health organization (WHO) of endocrine tumours. Endocr
Pathol. 28:213–227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lloyd RV, Osamura RY, Klöppel G and Rosai
J: WHO Classification of Tumours of Endocrine Organs. 4th. IACR;
Lyon: 2017
|
|
5
|
Roman-Gonzalez A and Jimenez C: Malignant
pheochromocytoma-paraganglioma: Pathogenesis, TNM staging and
current clinical trials. Curr Opin Endocrinol Diabetes Obes.
24:174–183. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amin B, Edge S and Greene F: AJCC cancer
staging manual. Springer; New York: 2017, View Article : Google Scholar
|
|
7
|
Lenders JW, Duh QY, Eisenhofer G,
Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K and
Young WF Jr; Endocrine Society, : Pheochromocytoma and
paraganglioma: An endocrine society clinical practice guideline. J
Clin Endocrinol Metab. 99:1915–1942. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Crona J, Taïeb D and Pacak K: New
perspectives on pheochromocytoma and paraganglioma: Towards a
molecular classification. Endocr Rev. 38:489–515. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pacak K and Del Rivero J: Pheochromocytoma
(Updated June 10, 2013) = Endotext. De Groot LJ, Chrousos G, Dungan
K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M,
McLachlan R, New M, et al: MDText.com, Inc.; South Dartmouth, MA:
2000
|
|
10
|
Neumann HP, Bausch B, McWhinney SR, Bender
BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K,
et al: Germ-line mutations in nonsyndromic pheochromocytoma. N Engl
J Med. 346:1459–1466. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pacak K and Wimalawansa SJ:
Pheochromocytoma and paraganglioma. Endocr Pract. 21:406–412. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qin Y, Yao L, King EE, Buddavarapu K,
Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F,
et al: Germline mutations in TMEM127 confer susceptibility to
pheochromocytoma. Nat Genet. 42:229–233. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsirlin A, Oo Y, Sharma R, Kansara A,
Gliwa A and Banerji MA: Pheochromocytoma: A review. Maturitas.
77:229–238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baysal BE, Ferrell RE, Willett-Brozick JE,
Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE,
Rubinstein WS, Myers EN, et al: Mutations in SDHD, a mitochondrial
complex II gene, in hereditary paraganglioma. Science. 287:848–851.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Niemann S and Müller U: Mutations in SDHC
cause autosomal dominant paraganglioma, type 3. Nat Genet.
26:268–270. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Astuti D, Latif F, Dallol A, Dahia PL,
Douglas F, George E, Sköldberg F, Husebye ES, Eng C and Maher ER:
Gene mutations in the succinate dehydrogenase subunit SDHB cause
susceptibility to familial pheochromocytoma and to familial
paraganglioma. Am J Hum Genet. 69:49–54. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Karasek D, Shah U, Frysak Z, Stratakis C
and Pacak K: An update on the genetics of pheochromocytoma. J Hum
Hypertens. 27:141–147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brouwers FM, Eisenhofer G, Tao JJ, Kant
JA, Adams KT, Linehan WM and Pacak K: High frequency of SDHB
germline mutations in patients with malignant
catecholamine-producing paragangliomas: Implications for genetic
testing. J Clin Endocrinol Metab. 91:4505–4509. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fishbein L, Leshchiner I, Walter V,
Danilova L, Robertson AG, Johnson AR, Lichtenberg TM, Murray BA,
Ghayee HK, Else T, Ling S, et al: Comprehensive molecular
characterization of pheochromocytoma and paraganglioma. Cancer
Cell. 31:181–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
NGS, in PPGL (NGSnPPGL) Study Group, ;
Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, Welander J,
Tops CM, Firth H, Dwight T, et al: Consensus statement on
next-generation-sequencing-based diagnostic testing of hereditary
phaeochromocytomas and paragangliomas. Nat Rev Endocrinol.
13:233–247. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zelinka T and Widimský J:
Pheochromocytoma-why is its early diagnosis so important for
patient? Vnitr Lek. 61:487–491. 2015.PubMed/NCBI
|
|
22
|
Pacak K, Linehan WM, Eisenhofer G, Walther
MM and Goldstein DS: Recent advances in genetics, diagnosis,
localization and treatment of pheochromocytoma. Ann Intern Med.
134:315–329. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen H, Sippel RS, O'Dorisio MS, Vinik AI,
Lloyd RV and Pacak K; North American Neuroendocrine Tumor Society
(NANETS), : The North American neuroendocrine tumor society
consensus guideline for the diagnosis and management of
neuroendocrine tumors: Pheochromocytoma, paraganglioma and
medullary thyroid cancer. Pancreas. 39:775–783. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lenders JW, Pacak K, Walther MM, Linehan
WM, Mannelli M, Friberg P, Keiser HR, Goldstein DS and Eisenhofer
G: Biochemical diagnosis of pheochromocytoma: Which test is best?
JAMA. 287:1427–1434. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eisenhofer G, Siegert G, Kotzerke J,
Bornstein SR and Pacak K: Current progress and future challenges in
the biochemical diagnosis and treatment of pheochromocytomas and
paragangliomas. Horm Metab Res. 40:329–337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eisenhofer G, Goldstein DS, Walther MM,
Friberg P, Lenders JW, Keiser HR and Pacak K: Biochemical diagnosis
of pheochromocytoma: How to distinguish true-from false-positive
test results. J Clin Endocrinol Metab. 88:2656–2666. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jacques AE, Sahdev A, Sandrasagara M,
Goldstein R, Berney D, Rockall AG, Chew S and Reznek RH: Adrenal
phaeochromocytoma: Correlation of MRI appearances with histology
and function. Eur Radiol. 18:2885–2892. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Motta-Ramirez GA, Remer EM, Herts BR, Gill
IS and Hamrahian AH: Comparison of CT Findings in symptomatic and
incidentally discovered pheochromocytomas. Am J Roentgenol.
185:684–688. 2005. View Article : Google Scholar
|
|
29
|
Čtvrtlík F, Heřman M, Študent V, Tichá V
and Minařík J: Differential diagnosis of incidentally detected
adrenal masses revealed on routine abdominal CT. Eur J Radiol.
69:243–252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Patel J, Davenport MS, Cohan RH and Caoili
EM: Can established CT attenuation and washout criteria for adrenal
adenoma accurately exclude pheochromocytoma? AJR Am J Roentgenol.
201:122–127. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schieda N, Alrashed A, Flood TA, Samji K,
Shabana W and McInnes MD: Comparison of quantitative MRI and CT
washout analysis for differentiation of adrenal pheochromocytoma
from adrenal adenoma. AJR Am J Roentgenol. 206:1141–1148. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang GM, Shi B, Sun H, Jin ZY and Xue HD:
Differentiating pheochromocytoma from lipid-poor adrenocortical
adenoma by CT texture analysis: Feasibility study. Abdom Radiol.
42:2305–2313. 2017. View Article : Google Scholar
|
|
33
|
McDermott S, McCarthy CJ and Blake MA:
Images of pheochromocytoma in adrenal glands. Gland Surg.
4:350–358. 2015.PubMed/NCBI
|
|
34
|
Pacak K, Goldstein DS, Doppman JL, Shulkin
BL, Udelsman R and Eisenhofer G: A ‘pheo’ lurks: Novel approaches
for locating occult pheochromocytoma. J Clin Endocrinol Metab.
86:3641–3646. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Korobkin M, Giordano TJ, Brodeur FJ,
Francis IR, Siegelman ES, Quint LE, Dunnick NR, Heiken JP and Wang
HH: Adrenal adenomas: Relationship between histologic lipid and CT
and MR findings. Radiology. 200:743–747. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Remer EM, Motta-Ramirez GA, Shepardson LB,
Hamrahian AH and Herts BR: CT histogram analysis in pathologically
proven adrenal masses. Am J Roentgenol. 187:191–196. 2006.
View Article : Google Scholar
|
|
37
|
Lubner MG, Smith AD, Sandrasegaran K,
Sahani DV and Pickhardt PJ: CT texture analysis: Definitions,
applications, biologic correlates and challenges. RadioGraphics.
37:1483–1503. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Northcutt BG, Raman SP, Long C, Oshmyansky
AR, Siegelman SS, Fishman EK and Johnson PT: MDCT of adrenal
masses: Can dual-phase enhancement patterns be used to
differentiate adenoma and pheochromocytoma? AJR Am J Roentgenol.
201:834–839. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Korobkin M, Brodeur FJ, Yutzy GG, Francis
IR, Quint LE, Dunnick NR and Kazerooni EA: Differentiation of
adrenal adenomas from nonadenomas using CT attenuation values. AJR
Am J Roentgenol. 166:531–536. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Caoili EM, Korobkin M, Francis IR, Cohan
RH and Dunnick NR: Delayed enhanced CT of lipid-poor adrenal
adenomas. AJR Am J Roentgenol. 175:1411–1415. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Szolar DH, Korobkin M, Reittner P,
Berghold A, Bauernhofer T, Trummer H, Schoellnast H, Preidler KW
and Samonigg H: Adrenocortical carcinomas and adrenal
pheochromocytomas: Mass and enhancement loss evaluation at delayed
contrast-enhanced CT. Radiology. 234:479–485. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kumagae Y, Fukukura Y, Takumi K, Shindo T,
Tateyama A, Kamiyama T, Kamimura K and Nakajo M: Distinguishing
adrenal adenomas from non-adenomas on dynamic enhanced CT: A
comparison of 5 and 10 min delays after intravenous contrast medium
injection. Clin Radiol. 68:696–703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mukherjee JJ, Peppercorn PD, Reznek RH,
Patel V, Kaltsas G, Besser M and Grossman AB: Pheochromocytoma:
Effect of nonionic contrast medium in CT on circulating
catecholamine levels. Radiology. 202:227–231. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bessell-Browne R and O'Malley ME: CT of
pheochromocytoma and paraganglioma: Risk of adverse events with
i.v. administration of nonionic contrast material. AJR Am J
Roentgenol. 188:970–974. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mansmann G, Lau J, Balk E, Rothberg M,
Miyachi Y and Bornstein SR: The clinically inapparent adrenal mass:
Update in diagnosis and management. Endocr Rev. 25:309–340. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tsushima Y, Takahashi-Taketomi A and Endo
K: Diagnostic utility of diffusion-weighted MR imaging and apparent
diffusion coefficient value for the diagnosis of adrenal tumors. J
Magn Reson Imaging. 29:112–117. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Umanodan T, Fukukura Y, Kumagae Y, Shindo
T, Nakajo M, Takumi K, Nakajo M, Hakamada H, Umanodan A and
Yoshiura T: ADC histogram analysis for adrenal tumor histogram
analysis of apparent diffusion coefficient in differentiating
adrenal adenoma from pheochromocytoma. J Magn Reson Imaging.
45:1195–1203. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dong Y and Liu Q: Differentiation of
malignant from benign pheochromocytomas with diffusion-weighted and
dynamic contrast-enhanced magnetic resonance at 3.0 T. J Comput
Assist Tomogr. 36:361–366. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim S, Salibi N, Hardie AD, Xu J, Lim RP,
Lee VS and Taouli B: Characterization of adrenal pheochromocytoma
using respiratory-triggered proton MR spectroscopy: Initial
experience. AJR Am J Roentgenol. 192:450–454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Melo HJ, Goldman SM, Szejnfeld J, Faria
JF, Huayllas MK, Andreoni C and Kater CE: Application of a protocol
for magnetic resonance spectroscopy of adrenal glands: An
experiment with over 100 cases. Radiol Bras. 47:333–341. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Reimer P, Parizel PM and Stichnoth FA:
Clinical MR imaging: A practical approach Second, completely
revised and updated. Springer-Verlag; Berlin Heidelberg New York:
2006, View Article : Google Scholar
|
|
52
|
Shellock FG and Kanal E: Safety of
magnetic resonance imaging contrast agents. J Magn Reson Imaging.
10:477–484. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Taïeb D, Timmers HJ, Hindié E, Guillet BA,
Neumann HP, Walz MK, Opocher G, de Herder WW, Boedeker CC and de
Krijger RR: EANM 2012 guidelines for radionuclide imaging of
phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging.
39:1977–1995. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bombardieri E, Giammarile F, Aktolun C,
Baum RP, Bischof Delaloye A, Maffioli L, Moncayo R, Mortelmans L,
Pepe G, Reske SN, et al: 131I/123I-metaiodobenzylguanidine (mIBG)
scintigraphy: Procedure guidelines for tumour imaging. Eur J Nucl
Med Mol Imaging. 37:2436–2446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Treglia G, Cocciolillo F, de Waure C, Di
Nardo F, Gualano MR, Castaldi P, Rufini V and Giordano A:
Diagnostic performance of 18F-dihydroxyphenylalanine positron
emission tomography in patients with paraganglioma: A
meta-analysis. Eur J Nucl Med Mol Imaging. 39:1144–1153. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fiebrich HB, Brouwers AH, Kerstens MN,
Pijl ME, Kema IP, de Jong JR, Jager PL, Elsinga PH, Dierckx RA, van
der Wal JE, et al: 6-(F-18)Fluoro-l-dihydroxyphenylalanine positron
emission tomography is superior to conventional imaging with
123I-metaiodobenzylguanidine scintigraphy, computer tomography and
magnetic resonance imaging in localizing tumors causing
catecholamine excess. J Clin Endocrinol Metab. 94:3922–3930. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Timmers HJ, Chen CC, Carrasquillo JA,
Whatley M, Ling A, Eisenhofer G, King KS, Rao JU, Wesley RA, Adams
KT and Pacak K: Staging and functional characterization of
pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose
(18F-FDG) positron emission tomography. J Natl Cancer
Inst. 104:700–708. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Janssen I, Chen CC, Millo CM, Ling A,
Taieb D, Lin FI, Adams KT, Wolf KI, Herscovitch P, Fojo AT, et al:
PET/CT comparing (68)Ga-DOTATATE and other radiopharmaceuticals and
in comparison with CT/MRI for the localization of sporadic
metastatic pheochromocytoma and paraganglioma. Eur J Nucl Med Mol
Imaging. 43:1784–1791. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jing H, Li F, Wang L, Wang Z, Li W, Huo L
and Zhang J: Comparison of the 68Ga-DOTATATA PET/CT, FDG PET/CT and
MIBG SPECT/CT in the evaluation of suspected primary
pheochromocytomas and paragangliomas. Clin Nucl Med. 42:525–529.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chang CA, Pattison DA, Tothill RW, Kong G,
Akhurst TJ, Hicks RJ and Hofman MS: 68Ga-DOTATATE and
18F-FDG PET/CT in paraganglioma and pheochromocytoma:
Utility, patterns and heterogeneity. Cancer Imaging. 16:2016.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tato A, Orte L, Diz P, Quereda C and
Ortuno J: Malignant pheochromocytoma, still a therapeutic
challenge. Am J Hypertens. 10:479–481. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Strong VE, Kennedy T, Al-Ahmadie H, Tang
L, Coleman J, Fong Y, Brennan M and Ghossein RA: Prognostic
indicators of malignancy in adrenal pheochromocytomas: Clinical,
histopathologic and cell cycle/apoptosis gene expression analysis.
Surgery. 143:759–768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Thompson LD: Pheochromocytoma of the
adrenal gland scaled score (PASS) to separate benign from malignant
neoplasms: A clinicopathologic and immunophenotypic study of 100
cases. Am J Surg Pathol. 26:551–566. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mlika M, Kourda N, Zorgati MM, Bahri S,
Ben Ammar S and Zermani R: Prognostic value of Pheochromocytoma of
the adrenal gland scaled score (Pass score) tests to separate
benign from malignant neoplasms. Tunis Med. 91:209–215.
2013.PubMed/NCBI
|
|
65
|
de Wailly P, Oragano L, Radé F, Beaulieu
A, Arnault V, Levillain P and Kraimps JL: Malignant
pheochromocytoma: New malignancy criteria. Langenbecks Arch Surg.
397:239–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Duregon E, Volante M, Bollito E, Goia M,
Buttigliero C, Zaggia B, Berruti A, Scagliotti GV and Papotti M:
Pitfalls in the diagnosis of adrenocortical tumors: A lesson from
300 consultation cases. Hum Pathol. 46:1799–1807. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ctvrtlik F, Koranda P and Tichy T: Adrenal
disease: A clinical update and overview of imaging. Biomed Pap Med
Fac Univ Palacky Olomouc Czech Repub. 158:23–34. 2014.PubMed/NCBI
|
|
68
|
Hodin R, Lubitz C, Phitayakorn R and
Stephen A: Diagnosis and management of pheochromocytoma. Curr Probl
Surg. 51:151–187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Vanderveen KA, Thompson SM, Callstrom MR,
Young WF Jr, Grant CS, Farley DR, Richards ML and Thompson GB:
Biopsy of pheochromocytomas and paragangliomas: Potential for
disaster. Surgery. 146:1158–1166. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Brunaud L, Nguyen-Thi PL, Mirallie E,
Raffaelli M, Vriens M, Theveniaud PE, Boutami M, Finnerty BM,
Vorselaars WM, Rinkes IB, et al: Predictive factors for
postoperative morbidity after laparoscopic adrenalectomy for
pheochromocytoma: A multicenter retrospective analysis in 225
patients. Surg Endosc. 30:1051–1059. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Perel Y, Schlumberger M, Marguerite G,
Alos N, Revillon Y, Sommelet D, De Lumley L, Flamant F, Dyon JF,
Lutz P, et al: Pheochromocytoma and paraganglioma in children: A
report of 24 cases of the french society of pediatric oncology.
Pediatr Hematol Oncol. 14:413–422. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wang W, Li P, Wang Y, Wang Y, Ma Z, Wang
G, Gao J and Zhou H: Effectiveness and safety of laparoscopic
adrenalectomy of large pheochromocytoma: A prospective,
nonrandomized, controlled study. Am J Surg. 210:230–235. 2015.
View Article : Google Scholar : PubMed/NCBI
|