Introduction
Sepsis-induced central nervous system (CNS)
dysfunction is a potentially irreversible acute brain dysfunction;
as many as 71% of such patients go onto develop septic
encephalopathy (SE) (1). SE is
caused by systemic inflammation induced by the immune response to
lipopolysaccharide stimulation (LPS). The resident CNS macrophages,
microglia, are the primary target cells of LPS and it has been
demonstrated that sepsis induced by peripheral injection of LPS
activates microglial cells (2,3). The
pathogenesis of SE may be influenced by various elements, including
the local and systemic secretion of pro-inflammatory mediators,
activation of microglia, alterations in homeostasis,
neurotransmitter imbalances and the effects of sepsis-induced
peripheral multiple organ failure (1). It has also been demonstrated that the
systemic injection of LPS, either as a single or repeated
injection, impacts the cognitive competence of rats (4).
LPS-induced pro-inflammatory molecules are vital for
regulating the growth and dissemination of pathogens; however,
overproduction of LPS may induce sepsis syndrome, also known as
endotoxin shock (5). Prior exposure
to low doses of LPS cause the cell to become resilient to
subsequent LPS challenge; cells develop tolerance to endotoxins,
which is known as endotoxin tolerance (5,6).
Endotoxin tolerance has been observed in vitro and in
vivo, in animal models and in humans (5). It is also associated with changes in
particular regulatory events, including deficiencies in the myeloid
differentiation primary response 88 (MyD88) (7), reduced IL-1 receptor-associated kinase
(IRAK)4-MyD88 association (8),
suppressed IRAK1 activation (9), as
well as the upregulation of negative regulators, including IRAK-M
(10), suppressor of cytokine
signaling 1 (SOCS1) and SH2 domain containing
inositol-5-phosphatase (SHIP-1) (11). Furthermore, studies have indicated
that nuclear factor (NF)-kB subunits (12), as well as peroxisome
proliferator-activator receptor γ (13) are involved in the development of
endotoxin tolerance. It has also been demonstrated that SOCS1
affects a series of signaling pathways, including NF-κB (14), c-Jun N terminal kinase and p38
(15).
In CNS cells, including microglia, oligodendrocytes,
astrocytes and neurons, SOCS1 expression is induced by LPS,
interferon (IFN)-β or -γ, and interleukin (IL)-4 or −6. The effects
of SOCS1 on neurological results vary greatly depending on the
disease context and the neuroinflammatory microenvironment
(16). SOCS1−/− mice
develop multiple organ failures (17). Additionally, macrophages from
SOCS1−/− mice are hypersensitive to toll-like receptor
(TLR) ligands, as indicated by increases in the expression of
pro-inflammatory chemokines and cytokines (18). It has been hypothesized that SOCS1
may be used as a novel therapeutic strategy to treat patients with
multiple sclerosis (19) and
different types of cancer (20).
SOCS1 serves a vital role in modulating TLR-mediated responses and
may therefore be used as a therapeutic regulator in CNS
diseases.
However, the nature of the endotoxin tolerance that
modulates SE remains unclear. Indeed, to the best of our knowledge,
no experiments have addressed the effect of SOCS1 on microglia
during the development of endotoxin tolerance.
microRNAs (miRs) are small noncoding RNAs that
regulate multiple human biological processes via the
posttranslational control of mRNA expression (21). A number of miRs modulate endotoxin
tolerance (22,23). It has been demonstrated that miR-155
serves crucial roles in the immune response, glioma, B-cell
malignancies and hematopoiesis. The contribution of miR-155 to the
development of endotoxin tolerance has also been demonstrated in
humans and an endotoxin shock mouse model (24,25).
miR-155 targets and represses several downstream TLR4 mediators,
including tumor necrosis factor (TNF)-α, transcription factor PU.1,
SHIP1, SOCS1, TNF receptor-associated factor-6, IRAK1 and
interferon regulatory factor 5, highlighting miR-155 in the
development of endotoxin tolerance (24).
The present study measured the expression of SOCS1
and miR-155 in microglia and the murine cortex under states of
inflammation and endotoxin tolerance, and investigated the function
of miR-155 in modulating endotoxin tolerance in microglia. The
results of the current study demonstrated that endotoxin tolerance
induced the downregulation of miR-155 and upregulated the
expression of SOCS1, thus decreasing the production of
pro-inflammatory TNF-α. This suggests that miR-155 may be used to
regulate inflammation and endotoxin tolerance in microglia.
Materials and methods
Cell culture
BV2 cells (American Type Culture Collection,
Manassas, VA, USA) were cultured in Dulbecco's modified Eagle's
medium (DMEM)-F12 (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal calf serum (ScienCell Research
Laboratories, Inc., Carlsbad, CA, USA), in a 5% CO2
humidified atmosphere at 37°C. Primary mouse (C57BL/6; Experimental
Animal Center of Nantong University, Nantong, China) microglial
cultures were cultured following a previously described protocol
(26).
Cell transfections
BV2 cell transfections of 200 nM miR-155 mimic
(UUAAUGCUAAUUGUGAUAGGGGU, double strand), miR-155 inhibitor
(AAUUACGAUUAACACUAUCCCCA, single strand), mimic negative control
(NC) (UUCUCCGAACGUGUCACGU, double strand) and inhibitor negative
control (AAGAGGCUUGCACAGUGCA, single strand; all Guangzhou RiboBio
Co., Ltd., Guangzhou, China) were performed using HiPerFect
Transfection Reagent (Qiagen, Inc., Valencia, CA, USA) following
the manufacturer's protocol. The following day, the medium was
changed and cells were subjected to single LPS (defined as LPS) or
repeated LPS (defined as LPS/LPS) challenge.
Cell tolerance model
To induce endotoxin tolerance, BV2 cells were
treated with 10 ng/ml LPS (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) for 18 h, followed by washout of LPS with PBS. Cells were
rested for a further 2 h prior to restimulation with 100 ng/ml LPS
for 4, 6, 8 or 10 h. To induce inflammation, BV2 cells were treated
with a single stimulation of 100 ng/ml LPS for 4 or 8 h (prior to
mRNA detection), or for 6 or 10 h (for protein detection). RNA,
protein and cell-free supernatants were collected at indicated
times, following the final stimulation, respectively.
Murine tolerance model
A total of 28 C57BL/6 mice (weighing 22–25 g; 4
mice/group; 14 males and 14 females) were obtained from
Experimental Animal Center of Nantong University, given access to
regular chow and sterile water at regaular intervals, and housed at
21–23°C with relative humidity of 50±5% on a 12 h light cycle. To
induce endotoxin tolerance, the C57BL/6 mice were injected
intraperitoneally (i.p.) with an initial dose of 5 mg/kg LPS. After
3 days, mice were re-challenged with 10 mg/kg LPS (i.p.) on day 1,
3 and 7. To induce inflammation, mice received an i.p. injection of
200 µl PBS 3 days prior to 10 mg/kg LPS administration for 1, 3 and
7 days. Mice were sacrificed on the day of their respective final
injections. Tissues were collected and stored at −80°C until use.
All animal experiments were performed in accordance with the Guide
for the Care and Use of Laboratory Animals (27) and were approved by the Ethics
Committee of the Nantong University (Nantong, China).
SOCS1 knockdown
Non-sense short interfering (sh)RNA (scramble),
SOCS1 shRNA1 and SOCS1 shRNA2 (Table
I) were designed by using the Sigma online database (https://www.sigmaaldrich.com/china-mainland/zh/life-science/functional-genomics-and-rnai/shrna/individual-genes.html)
of validated shRNAs. shRNAs (1 µg) were subcloned between the
BamHI and NotI site of the pGreenPuro shRNA
Expression Lentivector (System Biosciences, Palo Alto, CA, USA).
BV2 cells were transfected into the plasmid using the HiPerFect
Transfection Reagent following the manufacturer's protocol and 12 h
after transfection, cells were subjected to LPS or LPS/LPS
treatment.
 | Table I.shRNA sequences. |
Table I.
shRNA sequences.
| Gene | Sequences
(5′-3′) |
|---|
| SOCS1 shRNA1 | F:
gatccGCGCGACAGTCGCCAACGGAATTCAAGAGATTCCGTTGGCGACTGTCGCGCTTTTTg |
|
| R:
aattCAAAAACACCCTGCGGAACTTGTTCAATCTCTTGAATTGAACAAGTTCCGCAGGGTGg |
| SOCS1 shRNA2 | F:
gatccGCGAGACCTTCGACTGCCTTTTTCAAGAGAAAAGGCAGTCGAAGGTCTCGCTTTTTg |
|
| R:
aattCAAAAAGCGAGACCTTCGACTGCCTTTTCTCTTGAAAAAGGCAGTCGAAGGTCTCGCg |
| Scramble shRNA | F:
gatccTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTg |
|
| R:
aattCAAAAATTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAAg |
RNA isolation and reverse
transfection-quantitative polymerase chain reaction (RT-qPCR)
Total RNAs were extracted from cell cultures and
mice tissues using TRIzol (Thermo Fisher Scientific, Inc.). cDNAs
were synthesized using M-MuLV Reverse Transcriptase in the
QuantiTect® Reverse Transcription kit (Qiagen, Inc.) for
15 min at 42°C. qPCR was performed on a CFX96 Touch™
System Real-Time PCR Detection system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), using SYBR-Green (Takara, Bio, Inc., Otsu,
Japan). The thermocycling conditions were as follows: 40 Cycles of
denaturation at 94°C for 15 sec, annealing at 60°C for 15 sec and
extension at 72°C for 10 sec. The relative amount of transcripts
was calculated using the 2−ΔΔCq method (28). GAPDH and RNU6-1 were used as
housekeeping genes. SOCS1 was normalized to GAPDH and miR-155 was
normalized to RNU6-1. All primers used for the RT-qPCR analysis are
listed in Table II.
| Table II.Primer sequences. |
Table II.
Primer sequences.
| Gene | Sequences
(5′-3′) |
|---|
| SOCS1 | Forward:
GAGACCTTCGACTGCCTTTTC |
|
| Reverse:
TAGTCACGGAGTACCGGGTTAAG |
| GAPDH | Forward:
TGACCTCAACTACATGGTCTACA |
|
| Reverse:
CTTCCCATTCTCGGCCTTG |
| miR-155 | RT:
GTCGTATCCAGTGCAGGGTCCGAGG |
|
|
TATTCGCACTGGATACGACACCCCT |
|
| Forward:
CGCCTGTTAATGCTAATTGTGA |
|
| Reverse:
AGTGCAGGGTCCGAGGTAT |
| RNU6-1 | RT:
AACGCTTCACGAATTTGCGT |
|
| Forward:
CTCGCTTCGGCAGCACA |
|
| Reverse:
AACGCTTCACGAATTTGCGT |
Western blotting
Cells were lysed in a lysis buffer (20 mM Hepes, 120
mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1%
SDS, protease inhibitor). Cellular debris was removed by
centrifugation at 15,000 × g at 4°C for 30 min. The protein
concentrations in each sample were determined using NanoDrop 1000
(Thermo Fisher Scientific, Inc.). A total of 80 µg/lane were
resolved on 10% SDS-PAGE gels, transferred onto the polyvinylidene
difluoride membrane and blocked in 5% fat-free milk at room
temperature for 2 h. Membranes were then immunoblotted with
anti-SOCS1 (1:500; cat. no. ab9870; Abcam, Cambridge, UK) or
anti-β-actin (1:4,000; cat. no. 4970; Cell Signaling Technology,
Inc., Danvars, MA, USA) antibodies at 4°C overnight. Subsequently,
membranes were washed three times with PBS for 5 min each wash and
incubated with anti-goat (1:2,000; cat. no. sc-2020; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) or anti-rabbit (1:5,000; cat.
no. 7074; both Cell Signaling Technology, Inc.) horseradish
peroxidase-conjugated immunoglobulin G at 4°C for 2 h. Blots were
developed using an enhanced chemiluminescence kit (EMD Millipore,
Billerica, MA, USA). The density of bands was measured using ImageJ
1.42q software (National Institutes of Health, Bethesda, MD,
USA).
Immunocytochemistry
BV2 cells were fixed on coverslips with 4%
paraformaldehyde for 0.5 h at room temperature, followed by
membrane permeabilization using 0.1% Triton X-100. Cells were then
blocked with 5% bovine serum albumin (Sigma-Aldrich; Merck KGaA) at
room temperature for 2 h, and incubated with primary antibodies
against SOCS1 (1:100; cat. no. ab9870; Abcam) and cluster of
differentiation 11b (1:100; cat. no. ab128797; Abcam) at 4°C
overnight. Cells were then incubated with anti-goat immunoglobulin
(Ig)G conjugated with tetramethylrhodamine (1:2,000; cat. no.
ab6882; Abcam) or anti-rabbit IgG conjugated with fluorescein
isothiocyanate secondary antibodies (1:2,000; cat. no. ab6798;
Abcam) at 4°C for 2 h. Coverslips were mounted on a slide with
glycerol and cells were viewed using a confocal microscope.
Enzyme-linked immunosorbent assay
(ELISA)
BV2 were incubated for 24 h at 37°C in 5%
CO2, culture medium was collected and centrifuged at 700
× g at 4°C for 5 min. Cell-free supernatants were transferred to 96
well plate and TNF-α levels were measured using an ELISA kit (cat.
no. 560478; BD Biosciences, San Jose, CA, USA) following the
manufacturer's protocol.
Statistical analysis
Results were evaluated for statistical significance
using GraphPad Prism 5 (GraphPad software, Inc., La Jolla, CA,
USA). All data are presented as the mean ± standard error of the
mean. Two-way analysis of variance was performed, followed by
Turkey's HSD post hoc tests; these tests were used to determine
whether differences among groups were significant and P<0.05 was
determined to indicate a significant difference.
Results
The effects of inflammation and
endotoxin tolerance on miR-155 induction in murine microglia and
the mouse cortex
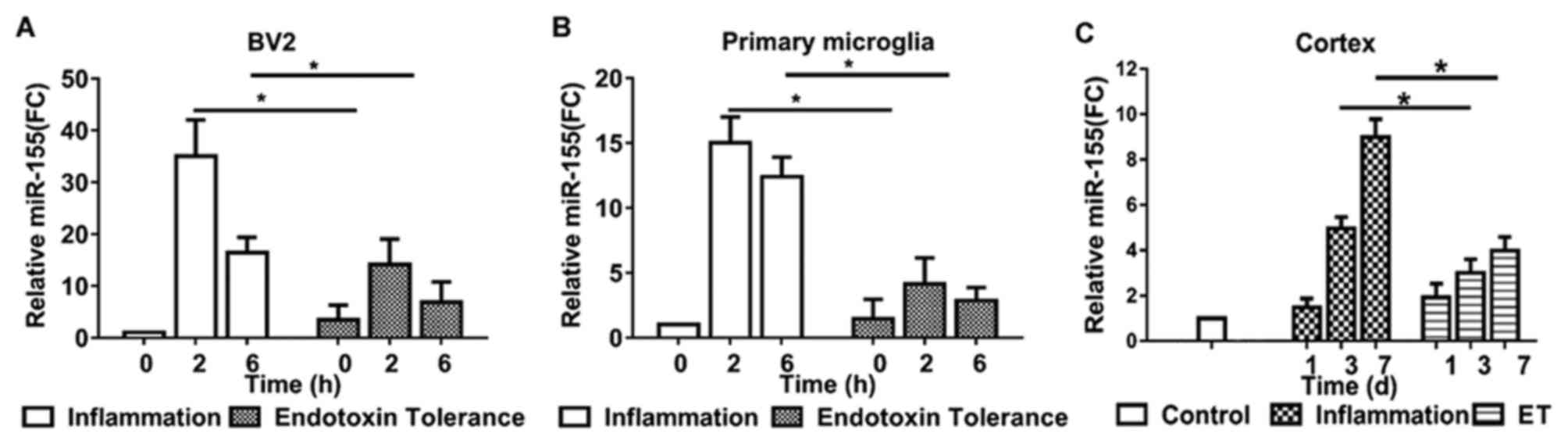
To investigate and compare miR-155 expression in
microglia during inflammation and endotoxin tolerance, BV2 cells
and primary microglia were treated with LPS to induce inflammation
or repeated LPS to induce endotoxin tolerance. miR-155 expression
was measured using RT-qPCR. miR-155 expression reached a peak at 2
h and subsequently declined in inflammatory BV2 (Fig. 1A) and primary microglia (Fig. 1B). Following treatment with repeated
LPS, cells became endotoxin tolerant; miR-155 expression did not
increase significantly following further stimulation in tolerant
BV2 (Fig. 1A) and primary microglia
(Fig. 1B).
To evaluate the expression of miR-155 in
inflammatory and endotoxin tolerant mouse microglia in vivo,
miR-155 levels in mouse cortex tissue were evaluated. Treatment of
mice with single LPS to induce inflammation produced similar
results to those of BV2 and primary microglia; mice exhibited a 4-
and 8-fold upregulation in miR-155 expression 3 and 7 days
following LPS administration, respectively. However, following
treatment of mice with repeated LPS to induce endotoxin tolerance,
miR-155 expression was only moderately upregulated in the 7 days
following subsequent LPS administration (Fig. 1C). These results indicate that
miR-155 serves a potential modulatory role during endotoxin
tolerance.
The effects of inflammation and
endotoxin tolerance on SOCS1 expression in murine microglia and
cortex
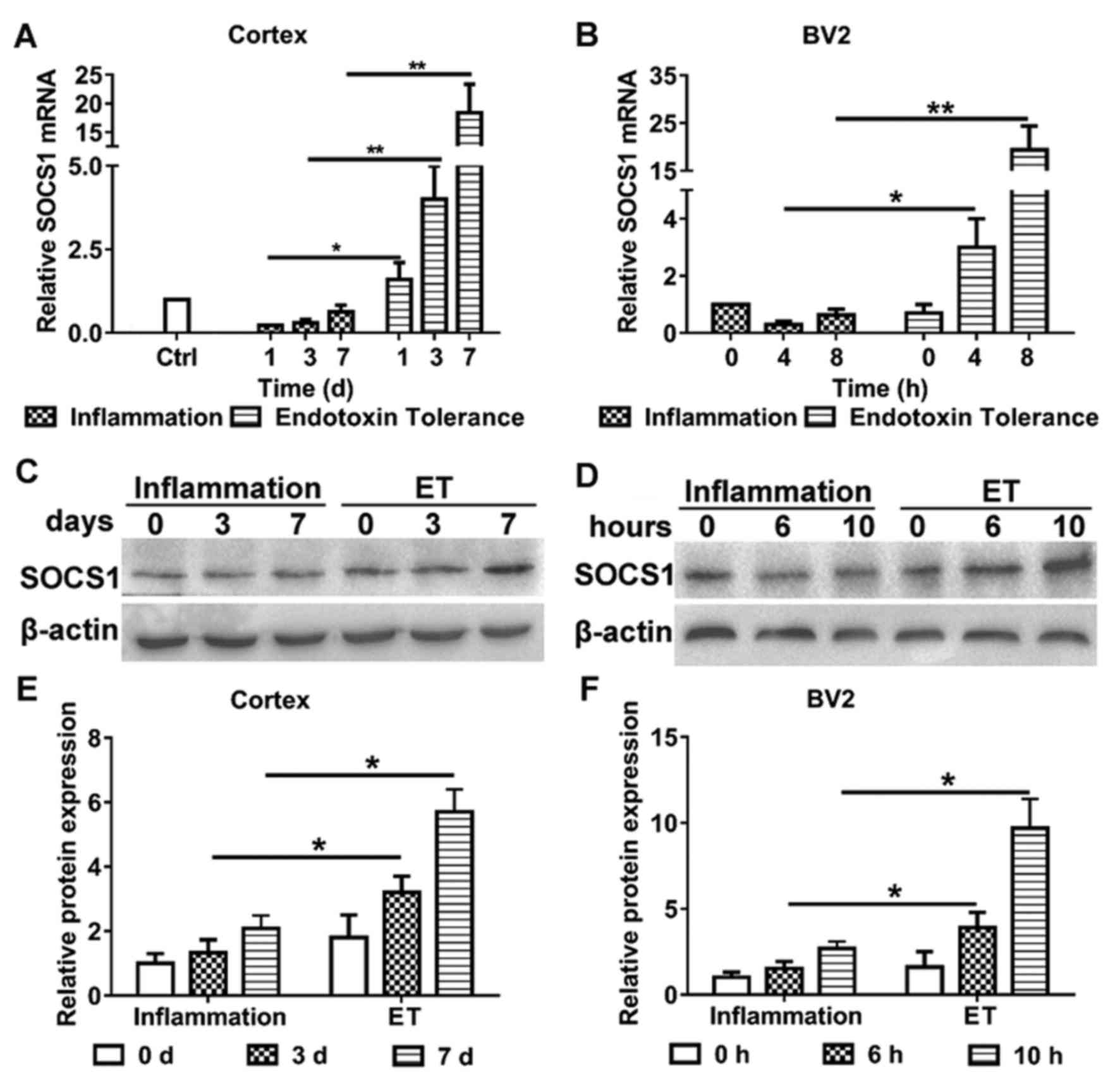
The aim of the present study was to clarify the role
of miR-155 in modulating endotoxin tolerance and whether it did
this by affecting SOCS1 expression. Therefore, the expression of
SOCS1 in murine microglial cells and the cortices of mice following
the induction of inflammation and endotoxin tolerance,
respectively, were evaluated (Fig.
2). Following the induction of endotoxin tolerance, the
expression of SOCS1 mRNA (Fig. 2A)
and protein (Fig. 2C and E) were
significantly increased in the mouse cortex, compared with mice
that only exhibited inflammation. Furthermore, BV2 cells treated
with repeated LPS exhibited increases in the expression of SOCS1
mRNA (Fig. 2B) and protein (Fig. 2D and F), compared with cells treated
with LPS alone. These results indicate that SOCS1 is involved in
modulating endotoxin tolerance in microglia.
Endotoxin tolerance mediates SOCS1
expression via miR-155 in murine microglia
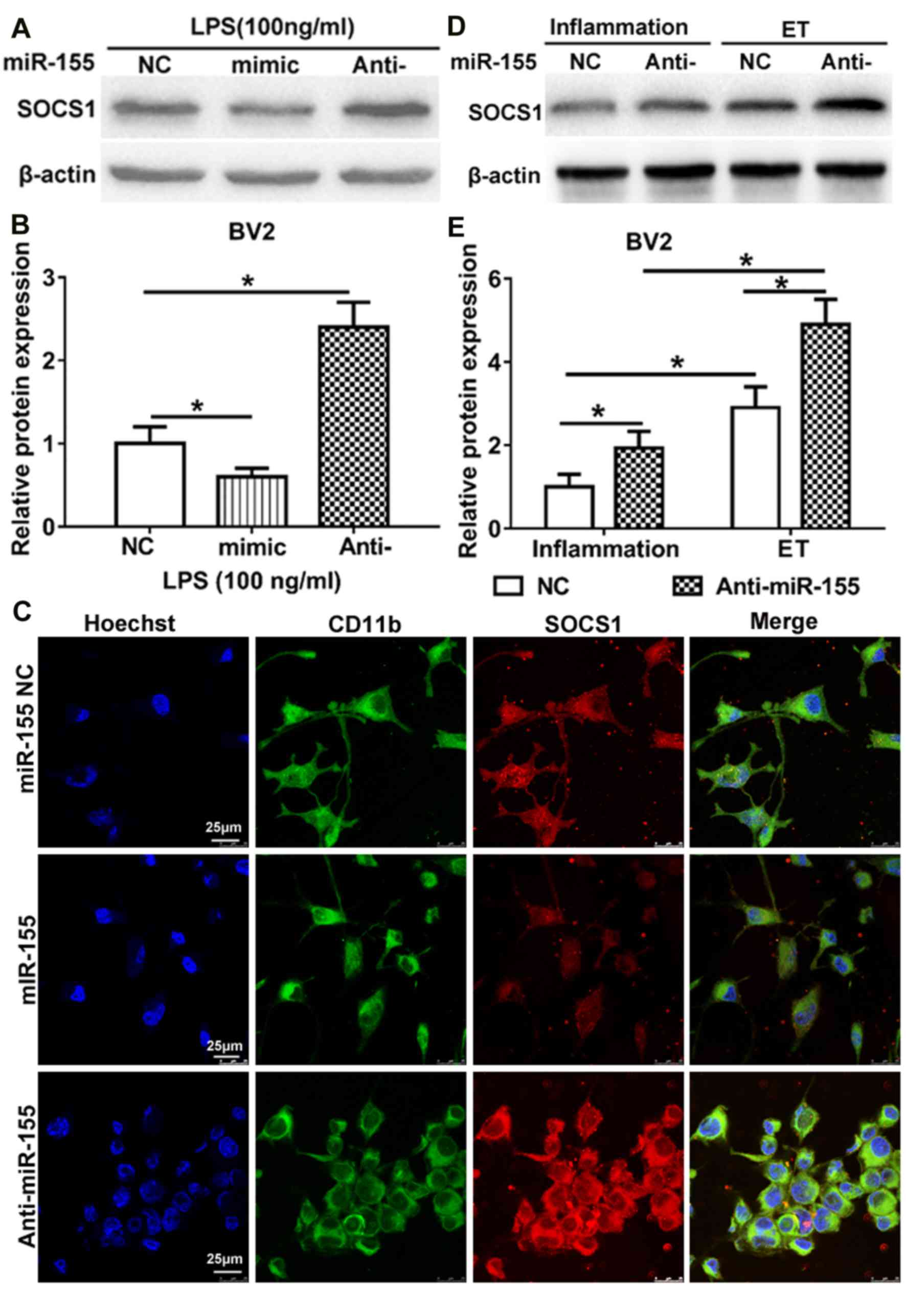
TargetScan indicated that miR-155 is able to bind to
the 3′-untranslated region of SOCS1 mRNA and previous studies have
demonstrated that miR-155 affects SOCS1 expression in LPS-treated
mouse N9 microglia and other immune cells (29–31). To
examine whether miR-155 regulates the expression of SOCS1 during
inflammation and endotoxin tolerance, the expression of SOCS1 in
LPS-treated BV2 microglia was measured. Compared with the NC, the
overexpression of miR-155 following transfection of miR-155 mimic
significantly downregulated SOCS1 expression, whereas inhibition of
miR-155 following transfection with miR-155 inhibitor significantly
upregulated SOCS1 expression (Fig. 3A
and B). The same results were identified following
immunocytochemistry to determine the cytoplasmic expression of
SOCS1 (Fig. 3C). These results are
partly in accordance with the results of a study by Cardoso et
al (31), which identified that
SOCS1 is a target of miR-155 in N9 microglia. These results
indicate that SOCS1 is a direct target of miR-155 in BV2 cells
following treatment with LPS.
To further investigate the role of miR-155 in
regulating the induction of endotoxin tolerance by SOCS1, miR-155
NC and miR-155 inhibitor were transfected in BV2 microglia prior to
the induction of inflammation and endotoxin tolerance. The results
demonstrated that the expression of SOCS1 was significantly
upregulated in BV2 microglia that were endotoxin tolerant compared
with those that had only undergone inflammation. This was the case
in BV2 microglia transfected with miR-155 inhibitor or the NC
(Fig. 3D and E). Furthermore, in BV2
microglia transfected with the miR-155 inhibitor, the expression of
SOCS1 was significantly increased following the induction of
endotoxin tolerance or inflammation, compared with those that had
undergone transfection with the NC. Taken together, these results
suggest that miR-155 inhibition maintains the state of endotoxin
tolerance in BV2 microglia, at least partly, via SOCS1.
The role of SOCS1 in anti-inflammatory
phenotype during endotoxin tolerance
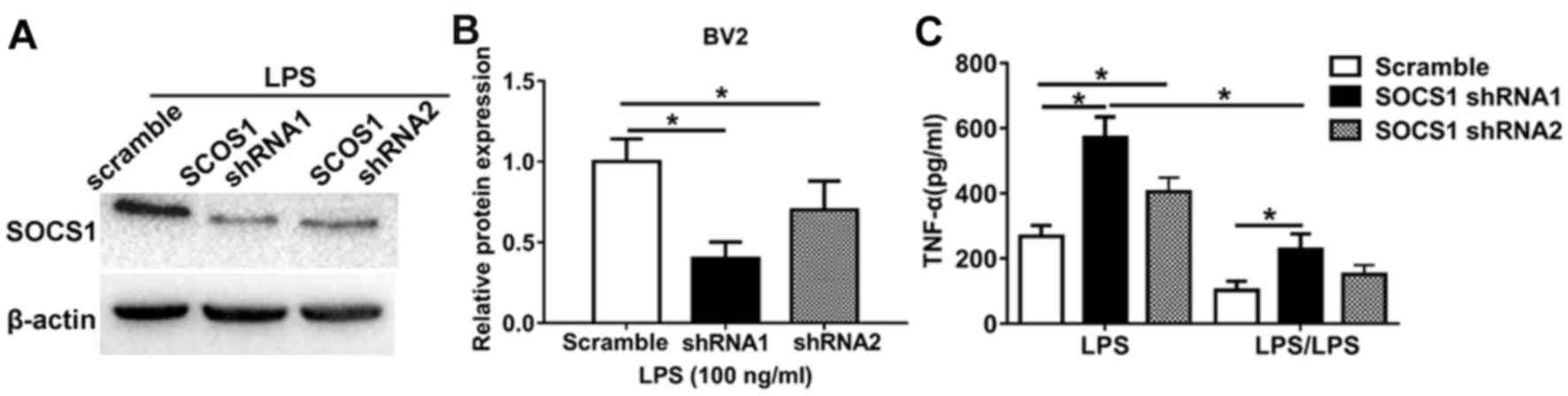
To further investigate the role of miR-155 in
regulating the SOCS1-induced microglia anti-inflammatory phenotype,
loss-of-function experiments were performed. Cells transfected with
an shRNA against SOCS1 was markedly (Fig. 4A) and significantly (Fig. 4B) low compared with cells transfected
with a scrambles shRNA. It was demonstrated that LPS significantly
increased TNF-α expression in cells transfected with SOCS1 shRNA1
and SOCS1 shRNA2 to knockdown SOCS1 expression compared with those
transfected with scramble shRNA cells (Fig. 4C). However, in cells treated with
LPS/LPS to induce endotoxin tolerance, the expression of TNF-α in
cells transfected with scramble shRNA was significantly decreased
compared with those transfected with SOCS1 shRNA. Notably, in cells
transfected with shRNA SOCS1 to induce SOCS1 knockdown, LPS
significantly increased TNF-α production compared with those
treated with LPS/LPS (Fig. 4C).
These data highlight the biological relevance of SOCS1 during the
development of endotoxin tolerance; silencing SOCS1 restores the
pro-inflammatory phenotype. Taken together, these results indicate
that miR-155 inhibition promotes the anti-inflammatory phenotype
and endotoxin tolerance in BV2 microglia and that this occurs by
increasing the expression of SOCS1.
Discussion
Endotoxin tolerance has been extensively
investigated in peripheral macrophages (5,7,9,10);
however, little is known about the microglia and miRs that are
involved in the development of endotoxin tolerance. Various studies
have identified that miR-155 serves an important in modulating the
innate and adaptive immune response; furthermore, the deregulation
of miR-155 has been implicated in a number of different diseases
(32,33). The present study demonstrated that
miR-155 serves a role in regulating the production of SOCS1 and
modulating endotoxin tolerance in BV2 microglia. This may improve
understanding regarding the pathogenetic mechanisms by which miR
regulates endotoxin tolerance.
A number of studies have demonstrated that SOCS1
serves important roles in the CNS (16). Several pathogens are able to induce
the expression of SOCS proteins as a method of evading the
IFN-mediated innate immune responses in CNS (15,16).
Pathogen-induced increases in SOCS expression in the CNS are
beneficial for the host (15,16).
Furthermore, SOCS1 influences the CNS inflammatory response during
infection (16). However, the
function of SOCS proteins in CNS immune cells has not yet been
investigated. The current study found that the expression of
miR-155 was upregulated and that the expression of SOCS1 was
downregulated in BV2 cells, primary microglia and mice cortices
following exposure to LPS. These results are in accordance with the
results of a previous study, which demonstrated that miR-155 was
upregulated and SOCS1 expression was decreased in N9 cells
(31).
miR-155 is processed from a non-coding transcript,
the B-cell Integration Cluster, and serves important functions in
cancer, the immune response and hematopoiesis (33). miR-155 expression is induced via TLRs
in dendritic cells and macrophages and markedly affects the
activities of these cells (29,30). In
microglia, the increase in miR-155 expression following activation
may stimulate the production of pro-inflammatory mediators
(34). It was demonstrated that
decreases in miR-155 expression significantly reduced the
expression of TNF-α and IL-6 (31).
Wen et al (35) demonstrated
that miR-155 promotes the inflammatory response by modulating
TLR4/SOCS1 expression in ischemic cerebral tissues. Previous
studies have also determined that miR-155 at least partly regulates
endotoxin sensitivity and tolerance in macrophages (24,36) and
alveolar epithelial cells (37) by
regulating SOCS1 expression. The results of the present study
illustrate that miR-155 expression is downregulated in BV2
microglia, primary microglia and mice cortices following exposure
to repeated LPS treatment. It was also demonstrated that SOCS1 mRNA
and protein levels increase in BV2 and murine cells that exhibit
endotoxin tolerance following repeated LPS treatment.
Interestingly, the changes in SOCS1 expression following repeated
LPS treatment were negatively associated with the change in miR-155
expression. Following transfection with miR-155 mimic and
inhibitor, it was demonstrated that miR-155 negatively regulates
SOCS1 expression in BV2 cells following exposure to LPS or repeated
LPS.
SOCS1 is a vital negative regulator of cytokines,
including TNF-α or IL-1β and therefore maintains the homeostasis of
the immune system (15–18). Although microglial activation
following pathological stimulation may be useful, it has been
demonstrated that the failure to inhibit microglia-mediated immune
responses at the appropriate time induces the overproduction of
inflammatory mediators and results in the development of a chronic
inflammatory state with severe consequences to ambient neurons
(38). The results of the current
study establish a direct link between miR-155 downregulation,
upregulated SOCS1 expression and the inhibition of pro-inflammatory
cytokines in BV2 cells following the development of endotoxin
tolerance. This suggests that miR-155 inhibition may contribute to
anti-inflammatory processes in the CNS by promoting the positive
function of SOCS1 and decreasing levels of pro-inflammatory
cytokines.
Taken together, the results of the current study
demonstrate that miR-155 inhibitors are able to downregulate BV2
microglia mediated neuroinflammation by regulating SOCS1 expression
and participate in sustaining endotoxin tolerance in CNS. These
results may facilitate the development of valuable therapeutic
strategies to treat CNS infections by decreasing inflammation and
endotoxin tolerance using miRs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81201252). This
project was also funded by the Scientific Research Programme of
Nantong (grant no. MS12016007).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
XSu, YQ and JS contributed to the conception and
design of experiments. XSh, JF and JY conducted the experiments.
XSu and YQ analyzed the data and drafted the manuscript. The final
version of the manuscript has been read and approved by all
authors, and each author believes that the manuscript represents
honest work.
Ethical approval
All experiments were performed in accordance with
the Guide for the Care and Use of Laboratory Animals and were
approved by the Ethics Committee of the Nantong University
(Nantong, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wilson JX and Young GB: Progress in
clinical neurosciences: Sepsis-associated encephalopathy: Evolving
concepts. Can J Neurol Sci. 30:98–105. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Buttini M, Limonta S and Boddeke HW:
Peripheral administration of lipopolysaccharide induces activation
of microglial cells in rat brain. Neurochem Int. 29:25–35. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nimmerjahn A, Kirchhoff F and Helmchen F:
Resting microglial cells are highly dynamic surveillants of brain
parenchyma in vivo. Science. 308:1314–1318. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sparkman NL, Kohman RA, Scott VJ and Boehm
GW: Bacterial endotoxin-induced behavioral alterations in two
variations of the Morris water maze. Physiol Behav. 86:244–251.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Biswas SK and Lopez-Collazo E: Endotoxin
tolerance: New mechanisms, molecules and clinical significance.
Trends Immunol. 30:475–487. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Foster SL, Hargreaves DC and Medzhitov R:
Gene-specific control of inflammation by TLR-induced chromatin
modifications. Nature. 447:972–978. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Medvedev AE, Lentschat A, Wahl LM,
Golenbock DT and Vogel SN: Dysregulation of LPS-induced Toll-like
receptor 4-MyD88 complex formation and IL-1 receptor-associated
kinase 1 activation in endotoxin-tolerant cells. J Immunol.
169:5209–5216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li L, Cousart S, Hu J and McCall CE:
Characterization of interleukin-1 receptor-associated kinase in
normal and endotoxin-tolerant cells. J Biol Chem. 275:23340–23345.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kobayashi K, Hernandez LD, Galán JE,
Janeway CA Jr, Medzhitov R and Flavell RA: IRAK-M is a negative
regulator of Toll-like receptor signaling. Cell. 110:191–202. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zacharioudaki V, Androulidaki A, Arranz A,
Vrentzos G, Margioris AN and Tsatsanis C: Adiponectin promotes
endotoxin tolerance in macrophages by inducing IRAK-M expression. J
Immunol. 182:6444–6451. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Piao W, Song C, Chen H, Diaz MA, Wahl LM,
Fitzgerald KA, Li L and Medvedev AE: Endotoxin tolerance
dysregulates MyD88- and Toll/IL-1R domain-containing adapter
inducing IFN-beta-dependent pathways and increases expression of
negative regulators of TLR signaling. J Leukoc Biol. 86:863–875.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Porta C, Rimoldi M, Raes G, Brys L, Ghezzi
P, Di Liberto D, Dieli F, Ghisletti S, Natoli G, De Baetselier P,
et al: Tolerance and M2 (alternative) macrophage polarization are
related processes orchestrated by p50 nuclear factor kappaB. Proc
Natl Acad Sci USA. 106:14978–14983. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Von Knethen AA and Brüne B: Delayed
activation of PPARgamma by LPS and IFN-gamma attenuates the
oxidative burst in macrophages. FASEB J. 15:535–544. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ryo A, Suizu F, Yoshida Y, Perrem K, Liou
YC, Wulf G, Rottapel R, Yamaoka S and Lu KP: Regulation of
NF-kappaB signaling by Pin1-dependent prolyl isomerization and
ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 12:1413–1426.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He Y, Zhang W, Zhang R, Zhang H and Min W:
SOCS1 inhibits tumor necrosis factor-induced activation of ASK1-JNK
inflammatory signaling by mediating ASK1 degradation. J Biol Chem.
281:5559–5566. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baker BJ, Akhtar LN and Benveniste EN:
SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol.
30:392–400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marine JC, Topham DJ, McKay C, Wang D,
Parganas E, Stravopodis D, Yoshimura A and Ihle JN: SOCS1
deficiency causes a lymphocyte-dependent perinatal lethality. Cell.
98:609–616. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gingras S, Parganas E, de Pauw A, Ihle JN
and Murray PJ: Re-examination of the role of suppressor of cytokine
signaling 1 (SOCS1) in the regulation of toll-like receptor
signaling. J Biol Chem. 279:54702–54707. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mujtaba MG, Flowers LO, Patel CB, Patel
RA, Haider MI and Johnson HM: Treatment of mice with the suppressor
of cytokine signaling-1 mimetic peptide, tyrosine kinase inhibitor
peptide, prevents development of the acute form of experimental
allergic encephalomyelitis and induces stable remission in the
chronic relapsing/remitting form. J Immunol. 175:5077–5086. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang FJ, Steeg PS, Price JE, Chiu WT,
Chou PC, Xie K, Sawaya R and Huang S: Molecular basis for the
critical role of suppressor of cytokine signaling-1 in melanoma
brain metastasis. Cancer Res. 68:9634–9642. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huntzinger E and Izaurralde E: Gene
silencing by microRNAs: Contributions of translational repression
and mRNA decay. Nat Rev Genet. 12:99–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Quinn EM, Wang J and Redmond HP: The
emerging role of microRNA in regulation of endotoxin tolerance. J
Leukoc Biol. 91:721–727. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nahid MA, Satoh M and Chan EK: MicroRNA in
TLR signaling and endotoxin tolerance. Cell Mol Immunol. 8:388–403.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Androulidaki A, Iliopoulos D, Arranz A,
Doxaki C, Schworer S, Zacharioudaki V, Margioris AN, Tsichlis PN
and Tsatsanis C: The kinase Akt1 controls macrophage response to
lipopolysaccharide by regulating microRNAs. Immunity. 31:220–231.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alexander M, Ramstead AG, Bauer KM, Lee
SH, Runtsch MC, Wallace J, Huffaker TB, Larsen DK, Tolmachova T,
Seabra MC, et al: Rab27-dependent exosome production inhibits
chronic inflammation and enables acute responses to inflammatory
stimuli. J Immunol. 199:3559–3570. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Saura J, Tusell JM and Serratosa J:
High-yield isolation of murine microglia by mild trypsinization.
Glia. 44:183–189. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
AnimalsGuide for the Care and Use of Laboratory Animals. 8th
edition. Washington (DC): National Academies Press (US). ISBN-13:
978-0-309-15400-0. 2011, PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
O'Connell RM, Chaudhuri AA, Rao DS and
Baltimore D: Inositol phosphatase SHIP1 is a primary target of
miR-155. Proc Natl Acad Sci USA. 106:7113–7118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
O'Connell RM, Taganov KD, Boldin MP, Cheng
G and Baltimore D: MicroRNA-155 is induced during the macrophage
inflammatory response. Proc Natl Acad Sci USA. 104:1604–1609. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cardoso AL, Guedes JR, de Almeida Pereira
L and de Lima Pedroso MC: miR-155 modulates microglia-mediated
immune response by down-regulating SOCS-1 and promoting cytokine
and nitric oxide production. Immunology. 135:73–88. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Staedel C and Darfeuille F: MicroRNAs and
bacterial infection. Cell Microbiol. 15:1496–1507. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
de Yébenes VG, Bartolomé-Izquierdo N and
Ramiro AR: Regulation of B-cell development and function by
microRNAs. Immunol Rev. 253:25–39. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kumar A, Stoica BA, Loane DJ, Yang M,
Abulwerdi G, Khan N, Kumar A, Thom SR and Faden AI:
Microglial-derived microparticles mediate neuroinflammation after
traumatic brain injury. J Neuroinflammation. 14:472017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wen Y, Zhang X, Dong L, Zhao J, Zhang C
and Zhu C: Acetylbritannilactone modulates MicroRNA-155-mediated
inflammatory response in ischemic cerebral tissues. Mol Med.
21:197–209. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Doxaki C, Kampranis SC, Eliopoulos AG,
Spilianakis C and Tsatsanis C: Coordinated regulation of miR-155
and miR-146a genes during induction of endotoxin tolerance in
macrophages. J Immunol. 195:5750–5761. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Neagos J, Standiford TJ, Newstead MW, Zeng
X, Huang SK and Ballinger MN: Epigenetic regulation of tolerance to
toll-like receptor ligands in alveolar epithelial cells. Am J
Respir Cell Mol Biol. 53:872–881. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Prinz M and Priller J: Microglia and brain
macrophages in the molecular age: From origin to neuropsychiatric
disease. Nat Rev Neurosci. 15:300–312. 2014. View Article : Google Scholar : PubMed/NCBI
|