Introduction
Familial hypercholesterolemia (FH) is an inherited
genetic disease characterized by hyperlipidemia, skin xanthomas on
extensor tendons and/or corneal arcus, and early onset of all forms
of atherosclerotic diseases including premature death secondary to
lifelong pathogenic elevations of serum cholesterol (1). FH exhibits an autosomal dominant
transmission pattern with ≥90% penetrance (1). It is typically divided into two main
phenotypes: Heterozygous FH (heFH) and homozygous FH (hoFH). hoFH
is less common than heFH and patients with hoFH often exhibit more
severe symptomatic phenotypes with higher levels of serum
cholesterol (2).
It has been reported that mutations in more than 3
genes, including low density lipoprotein receptor (LDLR),
apolipoprotein B-100 (APOB), proprotein convertase
subtilisin/kexin type 9 and LDL receptor adaptor protein 1, are
involved in the pathogenesis of FH (3–6).
Notably, LDLR mutations account for 85–90% of FH cases and
>1,700 variants of LDLR have been identified (7,8). The
human LDLR gene encodes an 860-amino-acid protein that
serves a significant role in the uptake and degradation of LDL by
the LDLR pathway (9). An in
vivo study by Anderson indicated that mutations in the
LDLR gene could cause dysfunction of the LDLR protein,
leading to the destruction of the LDLR pathway in vivo,
resulting in elevated serum cholesterol levels and premature
coronary artery disease (10).
It has previously been reported that the incidence
of LDLR mutations is ~70% in Chinese patients with FH.
However, these patients remain largely unidentified, particularly
in rural areas, due to patients and clinicians having little
knowledge about this disease and limited access to genetic testing
(11–13). A few cases of FH in China have been
diagnosed based primarily on clinical symptoms, but the phenotype
of FH is highly variable (3).
Different gene mutations and dosage effects of modifier genes may
lead to discrepancies in clinical manifestations and responses to
drugs (14–16). It is therefore important to
investigate the association between FH genotype and phenotype. In
the present study, a novel compound heterozygosis was identified by
direct polymerase chain reaction (PCR) sequencing of LDLR.
Laboratory tests and bioinformatics analysis were also conducted to
investigate the possible role of this mutation.
Subjects and methods
Study subjects
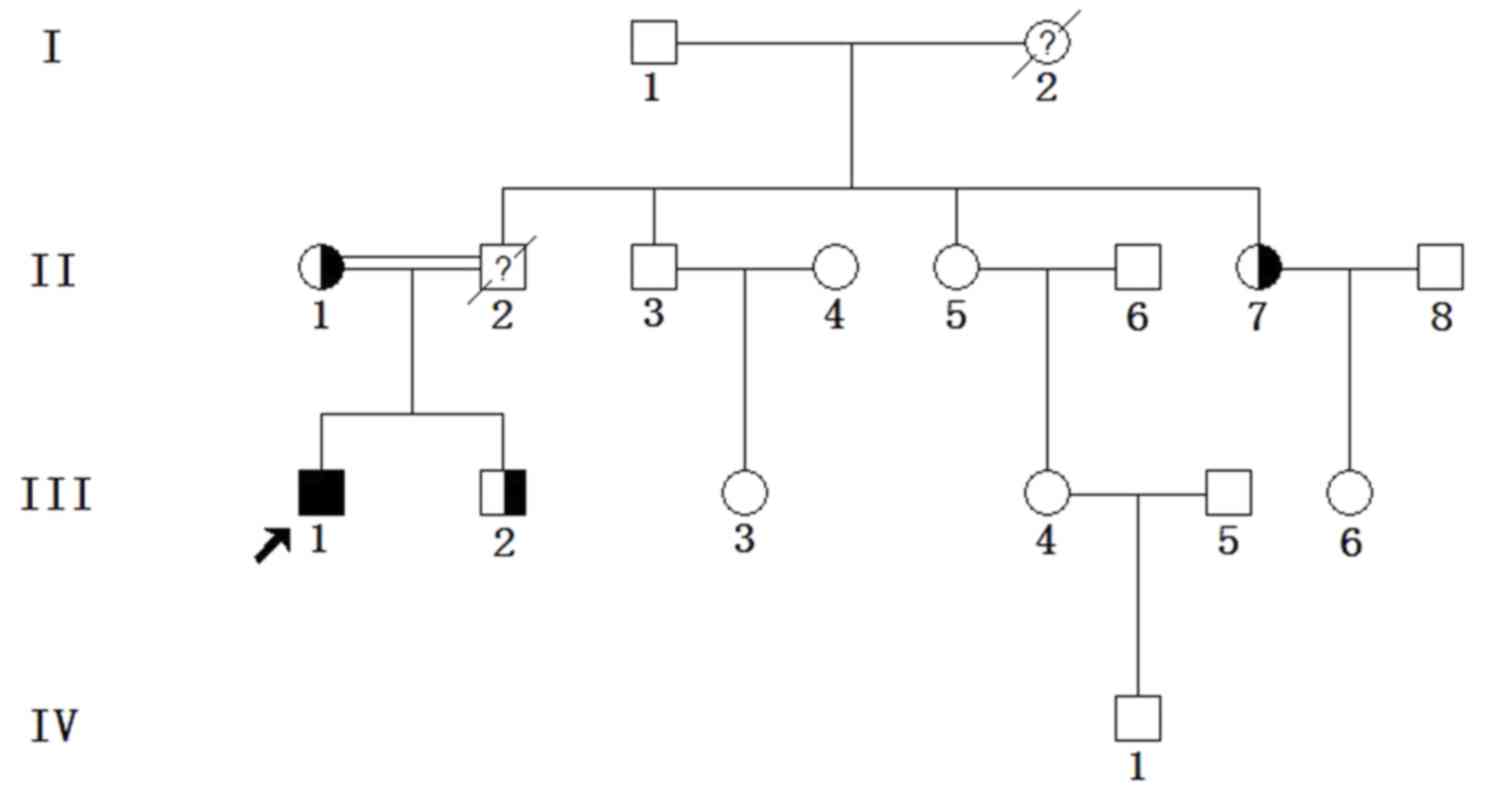
A total of 10 individuals from a Chinese FH family
were enrolled in the present study from December 2014 to May 2015
at Zhongnan Hospital of Wuhan University (Wuhan, China), including
4 males and 6 females, with an age range of 2–76 years (Fig. 1 and Table
I). All individuals enrolled in our study were members of this
FH family and consented to genetic and clinical examinations.
Individuals in the family who refused examinations were excluded.
The family history and basic physical information of subjects were
collected. The present study was approved by the Ethics Committee
of Zhongnan Hospital of Wuhan University. Prior informed consent
was obtained from all subjects, including the parents of
participants <16 years old. The young male proband (III1,
27-year-old) was the offspring of a consanguineous marriage (II1
and II2), as the grandfather of II1 and the grandmother of II2 were
siblings.
 | Table I.The serum lipid levels of the
subjects. |
Table I.
The serum lipid levels of the
subjects.
| Subjects | Sex | Age, years | TC mmol/l | TG mmol/l | HDL-C mmol/l | LDL-C mmol/l | APOA g/l | APOB g/l | LPa g/l | CRP mg/l | GLU mmol/l |
|---|
| Normal range | / | / | 2.8–5.8 | 0.45–1.81 | 0.9–2 | 2.1–3.3 | 1.05–1.75 | 0.6–1.4 | 0–0.3 | 0–8 | 3.8–6.1 |
| I1a | M | 76 | 4.50 | 0.81 | 1.08 | 2.14 | 1.52 | 0.85 | 0.14 | <0.5 | 3.90 |
| II1a | F | 52 | 8.10 | 1.14 | 1.04 | 5.02 | 1.37 | 1.72 | 0.48 | <0.5 | 4.30 |
| II5 | F | 48 | 5.10 | 0.75 | 1.45 | 2.38 | 1.89 | 0.89 | 0.09 | <0.5 | 3.90 |
| II7a | F | 45 | 9.40 | 1.53 | 1.65 | 4.93 | 2.17 | 1.86 | 0.52 | <0.5 | 3.90 |
| III1a | M | 27 | 18.30 | 2.67 | 0.60 | 11.60 | 1.11 | 3.81 | 0.14 | <0.5 | 3.50 |
| III2a | M | 22 | 8.20 | 0.92 | 1.05 | 4.86 | 1.46 | 1.65 | 0.06 | 6.46 | 4.50 |
| III3 | F | 22 | 3.90 | 0.61 | 1.34 | 1.61 | 1.77 | 0.61 | 0.13 | <0.5 | 4.00 |
| III4 | F | 27 | 4.50 | 0.33 | 1.58 | 1.84 | 1.85 | 0.66 | 0.27 | <0.5 | 4.00 |
| III6 | F | 22 | 3.90 | 0.74 | 1.13 | 1.69 | 1.64 | 0.68 | 0.48 | <0.5 | 3.90 |
| IV1 | M | 2 | 3.50 | 1.13 | 0.95 | 1.57 | 1.40 | 0.55 | 0.21 | <0.5 | 5.00 |
Clinical examinations and biochemical
tests
Blood samples were harvested following overnight
fasting. Serum was separated by centrifugation at 2,200 × g at 4°C
and examined using an automatic biochemical analyzer
(Abbot-AEROSET; Abbott Diagnostics, Santa Clara, CA, USA) for a
series of biochemical indices in the clinical laboratory department
of Zhongnan Hospital of Wuhan University: Blood glucose (GLU),
total cholesterol (TC), LDL-C, high-density lipoprotein cholesterol
(HDL-C), triglycerides, apolipoprotein (APO) A, APOB, lipoprotein a
and C-reactive protein.
All participants received
ultrasonography and electrocardiogram (EKG) examinations
A coronary contrast angiography was performed on the
proband using the digital subtract angiographic system (Phillip
Corp. FD 20; Phillips Healthcare, DA Best, The Netherlands). The
number and severity of peripheral artery atherosclerotic plaques
(PAS) of each individual was evaluated.
Genetic analysis
Genomic DNA was extracted from 1 ml peripheral blood
of all subjects using the sodium dodecyl sulfate-proteinase K
method as previously described (17). DNA quality was assessed using a
NanoDrop 2000c spectrophotometer (NanoDrop; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). All exons of LDLR were
screened for mutations using PCR direct sequencing as previously
described (18,19). Amplifications were performed in a
Hema 9600 PCR thermocycler (Hema Medical Instrument Co. Ltd,
Zhuhai, China) with a total volume of 25 µl, including 1× Taq DNA
Polymerase (Thermo Fisher Scientific, Inc.), 1× Taq Buffer, 0.5 µM
of forward and reverse primers (Table
II) and 100 ng DNA template. The conditions of PCR
amplification were 95°C for 30 sec for denaturation, annealing
temperature (Table II) for 45 sec
for annealing, 72°C for 45 sec for extension (38 cycles of
amplification). PCR products were evaluated by 2% agarose gel
electrophoresis, purified by an Axygen® PCR Clean-Up Kit
(Axygen Biosciences, Inc.; Corning Incorporated, Corning, NY, USA)
and directly sequenced using an ABI Genetic Analyzer 3730×1
(Applied Biosystems; Thermo Fisher Scientific, Inc.).
| Table II.Primers for the LDLR gene. |
Table II.
Primers for the LDLR gene.
| Exon | Forward primer
(5′→3′) | Reverse primer
(5′→3′) | Amplicon length
(bp) | Annealing
temperature (°C) |
|---|
| 5′ near
gene-Exon1 |
CTTCACCGGAGACCCAAATA |
TTCCCTTAAATCCCTCAGACTC | 592 | 58 |
| Exon2 |
CAGACTGTTCCTGATCGGATG |
AAGGGGTTAAGAATCGTGTCAC | 422 | 60 |
| Exon3 |
TGGGTCTTTCCTTTGAGTGAC |
TAGCACCATCCCCACTTTGT | 366 | 58 |
| Exon4 |
TAGAATGGGCTGGTGTTGGG |
TACTTTCTTGGCATGTTGTTGG | 567 | 60 |
| Exon5 |
AAGTAAGGTGGCACGATTATG |
AGCAGCAAGGCACAGAGAAT | 470 | 62 |
| Exon6 |
AAGCAAACTGAGGCTCAGACAC |
TGGAGTTCCCAAAACCCTACAG | 272 | 62 |
| Exon7 |
TGTAATGAGCCAAGGTTGGC |
GTTTGGTTGCCATGTCAGGAA | 261 | 58 |
| Exon8 |
GCTGTTTCCTTGATTACATCTC |
GATATGAGTCTGTGCAAAGTTC | 367 | 60 |
| Exon9-Exon10 |
CTTGGTTCCATCGACGGGTC |
CATGCCCAGCCCACTAACCA | 626 | 62 |
| Exon11 |
GGTTCCCAGCAGGACTATTTC |
GAAAGAGGGAAACCTTCAGG | 358 | 60 |
| Exon12 |
TGACCTCTCCTTATCCACTTGT |
CTCCTAGTCACAACCAGTTTTC | 272 | 60 |
| Exon13-Exon14 |
GAGGGTGGCCTGTGTCTCAT |
ATGAGTCCTTACAACGACCTTG | 605 | 60 |
| Exon15 |
GTCATTTGAGACTTTCGTCATTAG |
AAGAGGGCAAGAACTGTTATTAGAC | 457 | 60 |
| Exon16 |
CTGCCTGCTCCATTTCTTGG |
CTCCACATCCTCCATCTGACC | 349 | 60 |
| Exon17 |
TCAAGGTTATGGTACGATGCC |
TTGCCCTGTCACCATCTGAT | 485 | 62 |
| Exon18-3′ near
gene |
TTTCCTGAATGCTGGACTGAT |
GAGAAACTCAAAACTTCCTGGAG | 360 | 60 |
Multiple Sequence Alignments and
prediction of LDLR mutations
The alignment of LDLR protein sequences in multiple
species (obtained from the National Center for Biotechnology
Information protein database, www.ncbi.nlm.nih.gov/protein) was performed using
ClustalX2 software (Wellcome Trust Genome Campus, Hinxton, UK). The
secondary structure and hydrophobicity of the mutant LDLR protein
were predicted using ANTHEPROT 5.0 (PRABI-Lyon-Gerland, Lyon,
France), and three-dimensional (3D) structure prediction was
carried out using Swiss-Pdb Viewer 4.01 (Swiss Institute of
Bioinformatics, Lausanne, Switzerland) (20–22).
Results
FH pedigree
The young male proband (III1, 27-year-old) was the
offspring of a consanguineous marriage (II1 and II2), as the
grandfather of II1 and the grandmother of II2 were siblings
(Fig. 1). The proband was first
diagnosed with hypercholesterolemia in May 2015 when a biochemical
test was taken, due to aggravated angina. The proband presented
with multiple skin xanthomas (Fig.
2A) of various sizes on his extensor tendons in the fingers,
elbows, knees and Achilles. Notably, these xanthomas first appeared
when he was 10 years old. Arcus corneas, white rings in the corneal
margin, an indicator of lipid deposits, were found bilaterally
(Fig. 2A). The proband frequently
experienced (four times per day for two years) paroxysmal chest
discomfort and pain of several minutes duration each time, with
accompanying palpitations and breathlessness, which were heavier
during physical exertion. The proband's father (II2) died as a
result of lung cancer at the age of 33 and also had skin xanthomas
according to descriptions by other family members. The proband's
brother (III2, 22-year-old) exhibited moderate symptoms of partial
xanthoma and sporadic angina. Abnormal symptoms did not manifest in
other family members.
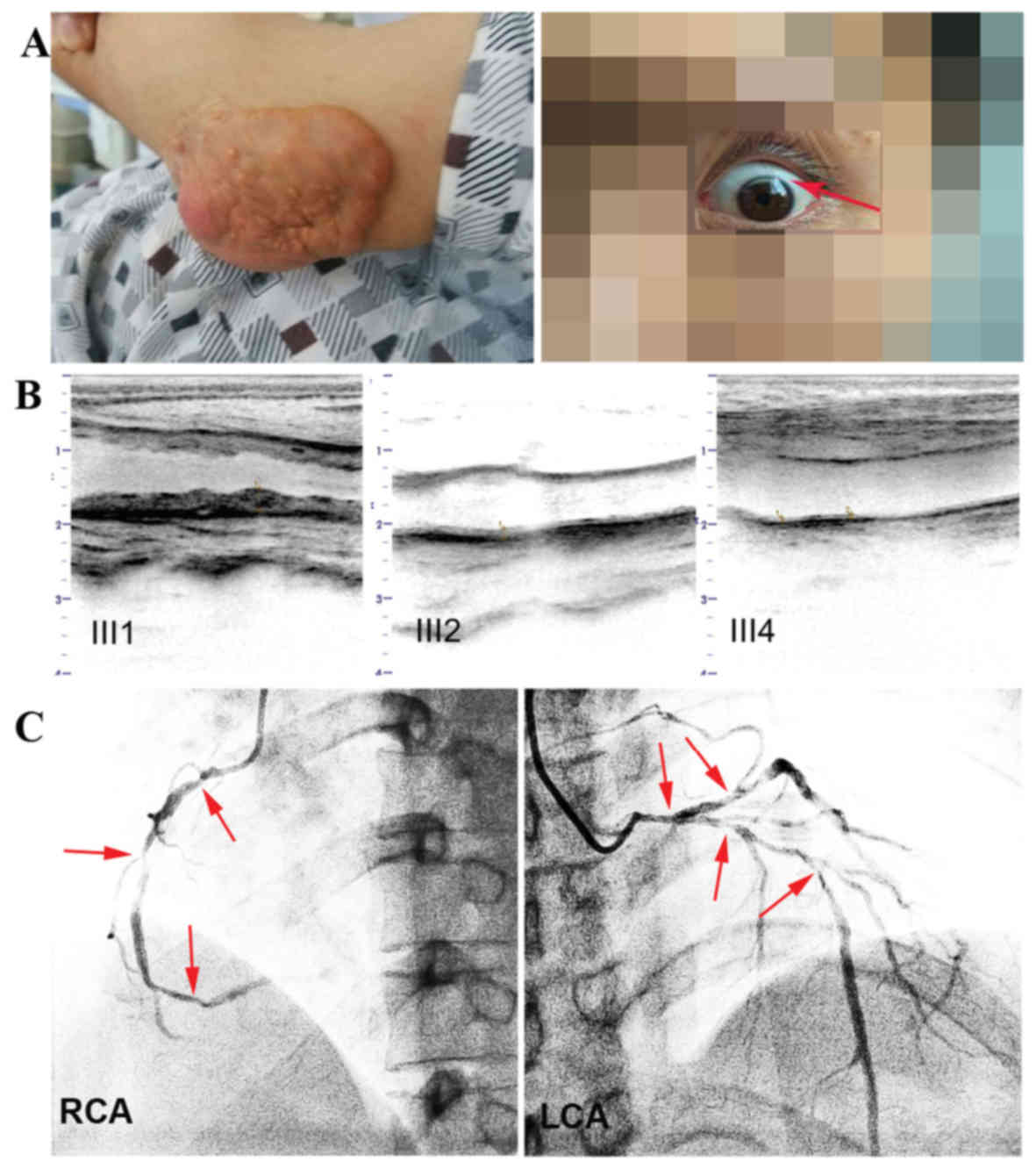 | Figure 2.Clinical characteristics of III1 and
photographic, ultrasonography results. (A) Elbow tendon xanthomata
and the corneal arcus, which is a shadow-like white curve on the
upper edge of the cornea. (B) Comparison of the distal carotid
segment atherosclerosis between patients III1, III2 and III4. (C)
Coronary angiogram from III1. Arrows indicate multiple stenosis of
the RCA and LCA. III1, the proband. RCA, right coronary artery;
LCA, left coronary artery. III1, the proband, the compound heFH
subject. III2, the proband's brother, an heFH subject. III4, the
proband's cousin, an unaffected subject. |
Results of clinical examinations and
biochemical tests
The blood lipid levels of the subjects were shown in
Table I. The proband exhibited
elevated levels of TC (18.30 mmol/l), LDL-C (11.60 mmol/l) and APOB
(3.81 g/l) compared with the normal range; the proband's mother,
brother and aunt, heterozygous in this study, also had high TC
(8.10–9.40 mmol/l), LDL-C (4.86–5.02 mmol/l) and APOB (1.65–1.86
g/l) levels (Table I). The rest of
the subjects were found to have levels of TC, LDL-C and APOB within
normal ranges (Table I).
Furthermore, HDL-C level in the proband (0.60 mmol/l) was lower
than the normal range, whereas it was within the normal range for
all other subjects. These data are consistent with the diagnostic
criteria for FH (23).
Clinical features and examination
results are listed in Table
III
For the proband, precordial auscultation revealed a
3–4/6 grade systolic murmur at the area of aortic valve. EKG found
that the ST segment depressed in V4, V5 and V6 leads reaching to
0.5 mv. Angiography revealed diffuse and heavy coronary artery
stenosis. The left main coronary trunk and the anterior descending
branch narrowed at 80% and the proximal circumflex branch at 50%;
the right coronary narrowed in the proximal segment at 85%, the
middle at 85%, and the distal at 70% (Fig. 2C). Ultrasonography revealed that the
aortic valve was calcified with mild stenosis and regurgitation,
the mitral valve had mild regurgitation, and there was extensive
and heavy peripheral arterial atherosclerosis (Fig. 2B). By contrast, other family members
presented only subtle abnormalities or normal manifestations.
Subjects with unique heterozygote mutations presented with mild
abnormalities. Ultrasonography revealed fewer plaques (II1, II7,
III2) and no cardiac structural malformation and dysfunction, and
the EKGs found mild ST segment depression (II1) or normal state.
The healthy members with no causative mutations (II5, III3, III4,
III6, IV1) had normal cardiac and peripheral arterial results, with
the exception of the proband's grandfather (I1) who had aortic
valve calcification and mild regurgitation as well as inferior Q
wave in the EKG. It was concluded that these characteristics were
most likely due to age (Table
III). Numerous peripheral artery atherosclerotic plaques were
observed in the proband, and five, two and three plaques were
observed in the proband's mother, brother and aunt, respectively.
However, atherosclerotic plaques were not observed in non-FH
members.
Trp557Term and Pro685Leu mutations in
LDLR gene
The results from the genetic analysis are presented
in Table III. Compound
heterozygote mutations (Trp577Term and Pro685Leu) in LDLR were
identified in the proband (Fig. 3A and
B). The proband's mother (II1) and brother (III2) were
heterozygous for the Trp577Term mutation and his aunt (II7) was
heterozygous for the Pro685Leu mutation.
Multiple sequence alignments and
prediction of LDLR mutations
Multiple sequence alignments revealed that these two
amino acid alterations (Trp577Term and Pro685Leu) were located in
the highly conserved region of LDLR in different species (Fig. 3C). The 3D ribbon model in silico
prediction identified an obvious truncation in mutant LDLR protein
caused by the Trp577Term mutation, which may lead to
haplo-insufficiency (Fig. 4A and B).
The secondary structure prediction revealed that the Pro685Leu
mutant LDLR appeared to have more β-strands in its secondary
structure than the wild type (Fig. 4C
and D). Furthermore, the hydrophobicity of the Pro685Leu mutant
protein was altered at the corresponding mutant region (Fig. 4E and F). Combined, these variations
in the physicochemical properties of LDLR may be responsible for
its functional abnormality.
Discussion
LDLR mutations are known to cause FH. In the
present study, two novel compound heterozygous LDLR
mutations, Trp577Term and Pro685Leu, were identified in the proband
of a Chinese FH family. Furthermore, one of these two mutations was
detected in the proband's mother, brother and aunt. According to
pedigree analysis (Fig. 1), the
Pro685Leu mutation was paternally inherited, whereas the Trp577Term
was maternally inherited. In this particular family, all mutation
carriers were found to have elevated serum lipid levels, typical
atherosclerotic plaques and coronary artery stenosis, all of which
are indicators of early onset atherosclerosis. Notably, these
symptoms were more severe in the compound heterozygote proband,
suggesting that the LDLR mutation has a possible dosage
effect in lipid metabolism. It was observed that the serum level of
HDL-C was decreased in the proband and within the normal range for
other family members, which suggests that elevated LDL-C may be
responsible for low HDL-C levels, resulting in a vicious cycle
effect in the proband. The proband also presented with bilateral
corneal arcus and non-ST segment elevation myocardial infarction,
and only he and his brother presented skin xanthomas. This
indicates a sex-biased phenotype (II1 vs. III2). There was little
difference observed in the phenotypes of two female suffers (II1
and II7), which suggests that the two mutations have a comparable
effect in FH. There was a step-wise severity in clinical symptoms
from hoFH, heFH to unaffected subjects, including xanthoma,
sporadic angina, hypercholesterolemia and peripheral artery
atherosclerotic plaques and stenosis. These results suggest a
genetic dosage-dependent clinical feature of FH.
The Trp577Term mutation is located in exon 12. The
typical outcome of this type of mutation is premature termination
of transcription, leading to a truncated protein lacking EGF,
O-linked sugars, membrane spanning and cytoplasmic structure
domains (9,24). The Pro685Leu mutation is located in
exon 14, which is the highly conserved region of the EGF precursor
domain in the LDLR protein (25).
Previous studies have demonstrated that this alteration affects the
flexibility of the LDLR polypeptide chain and the rigidity of the
peptide bonds adjacent to proline (26,27).
Based on previous reports, each of the Trp577Term
and Pro685Leu mutations in LDLR can cause FH independently
(26,28). The Trp577Term mutation has only been
documented in Chinese patients, and to the best of our knowledge,
the compound heterozygous mutations have not been reported
elsewhere. The present study indicated that compound heterozygous
mutations resulted in a severe clinical manifestation of FH.
In the present study, the proband's parents were
third degree relatives. It is well known that parents who both have
common heterozygous mutations in a consanguineous marriage tend to
give birth to homozygous children; however, in the present study
the proband was a compound heterozygote instead. It is therefore
worth studying the underlying association between incestuous family
histories and the incidence of heterozygous mutations. Two missense
mutations of Asn591Asn (c.1773C>T) and Val653Val (c.1959T>C)
were also identified in this family, which are also able to
increase LDL-C levels (29,30). However, the subjects carrying one or
both of these variations in the present study had normal levels of
TC and LDL-C. Since the study population of the above two studies
were Canadians and Japanese, respectively, we speculate that the
different outcome of these variations in our study may be
influenced by the difference of environmental or ethnic
factors.
There were certain limitations in the present study
that must be considered. In the present study, the mechanism of
destruction effects of Trp577Term and Pro685Leu mutations were only
analyzed using bioinformatics tools. Further biological studies are
required to confirm the underlying mechanism on mutant protein
functions. Investigations involving more families and more mutation
carriers will be helpful for the prevention and early intervention
for Chinese FH populations.
Acknowledgements
The authors of the present study would like to thank
the proband and his family for participating in this study, and
Enshi Center Hospital of China for their assistance.
Glossary
Abbreviations
Abbreviations:
|
FH
|
familial hypercholesterolemia
|
|
LDLR
|
low density lipoprotein receptor
|
|
PCR
|
polymerase chain reaction
|
|
heFH
|
heterozygous FH
|
|
hoFH
|
homozygous FH
|
|
APOB
|
apolipoprotein B
|
|
LDL-C
|
LDL cholesterol
|
|
GLU
|
blood glucose
|
|
TC
|
total cholesterol
|
|
HDL-C
|
high-density lipoprotein
cholesterol
|
|
EKG
|
electrocardiogram
|
References
|
1
|
Hopkins PN, Toth PP, Ballantyne CM and
Rader DJ; National Lipid Association Expert Panel on Familial
Hypercholesterolemia: Familial hypercholesterolemias: Prevalence,
genetics, diagnosis and screening recommendations from the National
Lipid Association Expert Panel on Familial Hypercholesterolemia. J
Clin Lipidol. 5(3 Suppl): S9–S17. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Raal FJ and Santos RD: Homozygous familial
hypercholesterolemia: Current perspectives on diagnosis and
treatment. Atherosclerosis. 223:262–268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van der Graaf A, Avis HJ, Kusters DM,
Vissers MN, Hutten BA, Defesche JC, Huijgen R, Fouchier SW, Wijburg
FA, Kastelein JJ and Wiegman A: Molecular basis of autosomal
dominant hypercholesterolemia: Assessment in a large cohort of
hypercholesterolemic children. Circulation. 123:1167–1173. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tada H, Kawashiri MA, Ohtani R, Noguchi T,
Nakanishi C, Konno T, Hayashi K, Nohara A, Inazu A, Kobayashi J, et
al: A novel type of familial hypercholesterolemia: Double
heterozygous mutations in LDL receptor and LDL receptor adaptor
protein 1 gene. Atherosclerosis. 219:663–666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mabuchi H, Nohara A, Noguchi T, Kobayashi
J, Kawashiri MA, Inoue T, Mori M, Tada H, Nakanishi C, Yagi K, et
al: Genotypic and phenotypic features in homozygous familial
hypercholesterolemia caused by proprotein convertase
subtilisin/kexin type 9 (PCSK9) gain-of-function mutation.
Atherosclerosis. 236:54–61. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rader DJ, Cohen J and Hobbs HH: Monogenic
hypercholesterolemia: New insights in pathogenesis and treatment. J
Clin Invest. 111:1795–1803. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldberg AC, Hopkins PN, Toth PP,
Ballantyne CM, Rader DJ, Robinson JG, Daniels SR, Gidding SS, de
Ferranti SD, Ito MK, et al: Familial hypercholesterolemia:
Screening, diagnosis and management of pediatric and adult
patients: Clinical guidance from the National Lipid Association
Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 5(3
Suppl): S1–S8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
University College London (UCL): LDLR FH
database. http://www.ucl.ac.uk/ldlr/Current/index.php?select_db=LDLRSeptember
14–2015
|
|
9
|
Soutar AK and Naoumova RP: Mechanisms of
disease: Genetic causes of familial hypercholesterolemia. Nat Clin
Pract Cardiovasc Med. 4:214–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anderson RG: Joe Goldstein and Mike Brown:
From cholesterol homeostasis to new paradigms in membrane biology.
Trends Cell Biol. 13:534–539. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mak YT, Pang CP, Tomlinson B, Zhang J,
Chan YS, Mak TW and Masarei JR: Mutations in the low-density
lipoprotein receptor gene in Chinese familial hypercholesterolemia
patients. Arterioscler Thromb Vasc Biol. 18:1600–1605. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shi Z, Yuan B, Zhao D, Taylor AW, Lin J
and Watts GF: Familial hypercholesterolemia in China: Prevalence
and evidence of underdetection and undertreatment in a community
population. Int J Cardiol. 174:834–836. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fan LL, Lin MJ, Chen YQ, Huang H, Peng DQ,
Xia K, Zhao SP and Xiang R: Novel mutations of low-density
lipoprotein receptor gene in china patients with familial
hypercholesterolemia. Appl Biochem Biotechnol. 176:101–109. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Desaphy JF, Gramegna G, Altamura C,
Dinardo MM, Imbrici P, George AL Jr, Modoni A, Lomonaco M and Conte
Camerino D: Functional characterization of ClC-1 mutations from
patients affected by recessive myotonia congenita presenting with
different clinical phenotypes. Exp Neurol. 248:530–540. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moosa MM, Ayub MI, Bashar AE, Sarwardi G,
Khan W, Khan H and Yeasmin S: Combination of two rare mutations
causes β-thalassaemia in a Bangladeshi patient. Genet Mol Biol.
34:406–409. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kasana BA, Dar WR, Aziz SA, Lone AR, Sofi
NU, Dar IA, Latief M, Arshad F and Hussain M and Hussain M:
Epidermal growth factor receptor mutation in adenocarcinoma lung in
a North Indian population: Prevalence and relation with different
clinical variables. Indian J Med Paediatr Oncol. 37:189–195. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Loparev VN, Cartas MA, Monken CE, Velpandi
A and Srinivasan A: An efficient and simple method of DNA
extraction from whole blood and cell lines to identify infectious
agents. J Virol Methods. 34:105–112. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng X, Ding J, Zheng F, Zhou X and Xiong
C: Two mutations in LDLR gene were found in two Chinese families
with familial hypercholesterolemia. Mol Biol Rep. 36:2053–2057.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li H, Zhang Y, Wei X, Peng Y, Yang P, Tan
H, Chen C, Pan Q, Liang D and Wu L: Rare intracranial cholesterol
deposition and a homozygous mutation of LDLR in a familial
hypercholesterolemia patient. Gene. 569:313–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mothobi ME, Guo S, Liu Y, Chen Q, Yussuf
AS, Zhu X and Fang Z: Mutation analysis of congenital cataract in a
Basotho family identified a new missense allele in CRYBB2. Mol Vis.
15:1470–1475. 2009.PubMed/NCBI
|
|
21
|
Chen Q, Ma J, Yan M, Mothobi ME, Liu Y and
Zheng F: A novel mutation in CRYAB associated with autosomal
dominant congenital nuclear cataract in a Chinese family. Mol Vis.
15:1359–1365. 2009.PubMed/NCBI
|
|
22
|
Yan M, Xiong C, Ye SQ, Chen Y, Ke M, Zheng
F and Zhou X: A novel connexin 50 (GJA8) mutation in a Chinese
family with a dominant congenital pulverulent nuclear cataract. Mol
Vis. 14:418–424. 2008.PubMed/NCBI
|
|
23
|
Hovingh GK, Davidson MH, Kastelein JJ and
O'Connor AM: Diagnosis and treatment of familial
hypercholesterolaemia. Eur Heart J. 34:962–971. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goldstein JL and Brown MS: The LDL
receptor. Arterioscler Thromb Vasc Biol. 29:431–438. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Davis CG, Goldstein JL, Südhof TC,
Anderson RG, Russell DW and Brown MS: Acid-dependent ligand
dissociation and recycling of LDL receptor mediated by growth
factor homology region. Nature. 326:760–765. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yao RE, Wang J, Geng J, Zheng Z, Yu T, Yu
Y and Fu Q: Identification of LDLR mutations in two Chinese
pedigrees with familial hypercholesterolemia. J Pediatr Endocrinol
Metab. 25:769–773. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun XM, Patel DD, Webb JC, Knight BL, Fan
LM, Cai HJ and Soutar AK: Familial hypercholesterolemia in China.
Identification of mutations in the LDL-receptor gene that result in
a receptor-negative phenotype. Arterioscler Thromb. 14:85–94. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hobbs HH, Brown MS and Goldstein JL:
Molecular genetics of the LDL receptor gene in familial
hypercholesterolemia. Hum Mutat. 1:445–466. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boright AP, Connelly PW, Brunt JH, Morgan
K and Hegele RA: Association and linkage of LDLR gene variation
with variation in plasma low density lipoprotein cholesterol. J Hum
Genet. 43:153–159. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamada Y, Ichihara S, Kato K, Yoshida T,
Yokoi K, Matsuo H, Watanabe S, Metoki N, Yoshida H, Satoh K, et al:
Genetic risk for metabolic syndrome: Examination of candidate gene
polymorphisms related to lipid metabolism in Japanese people. J Med
Genet. 45:22–28. 2008. View Article : Google Scholar : PubMed/NCBI
|