Introduction
Carotid atherosclerotic plaque rupture and secondary
thrombosis are two of the prominent causes of ischemic stroke
(1). Carotid artery lesions account
for 30% of ischemic strokes (2,3). Plaques
are divided into stable and vulnerable plaques (4). Vulnerable plaques refer to those that
rupture easily, are prothrombotic and unstable (5). When a vulnerable plaque ruptures and
becomes an embolus, the cerebral artery occludes, which eventually
leads to ischemic stroke (6). As a
result, it is of great clinical interest to stabilize vulnerable
plaques.
Usually, plaques prone to rupture are
morphologically characterized as having a thin fibrous cap
overlaying a large lipid core (7). A
fibrous cap primarily consists of vascular smooth muscle cells
(VSMCs) and the extracellular matrix (ECM) (8). The remodeling and transformation of the
ECM are important steps in the genesis and development of
atherosclerosis (9). Under normal
circumstances, a dynamic equilibrium exists between the generation
and degradation of the ECM (10).
Increasing evidence suggests that matrix metalloproteinases (MMPs)
are one of the main factors degrading the ECM, which causes the
rupture of plaques (11,12). MMPs belong to the calcium-dependent
zinc-containing protease superfamily (13). At present, >20 members of the MMP
family have been identified (14).
However, the roles that different MMPs serve in plaque stability
have not been extensively studied. For example, Graham et al
(15) revealed that the higher the
serum MMP-9 level, the more unstable the plaque, suggesting that
MMP-9 is associated with the plaque stability. Heo et al
(16) revealed that MMP-2 and MMP-9
were highly expressed in the ulcerated plaques, suggesting that
MMP-2 and MMP-9 may correlate with plaque rupture. In addition,
only a small amount of data exists that allows for the quantitative
and systematic evaluation of the expression of various types of
MMPs in human carotid plaques, and determines the key factors that
are associated with plaque vulnerability (17).
The vulnerability of plaques is a rather complicated
issue in which neovascularization and calcification factors are
involved (18,19). Calcification is a pivotal event in
the development of atherosclerosis (20,21).
During vascular calcification, VSMCs synthesize several osteogenic
factors, including osteopontin (OPN) and bone sialoprotein 2 (BSP)
(22,23). OPN and BSP are considered to be
important enzymes for the generation of calcium salt (24). However, the correlation between the
associated factors, including BSP and OPN, and plaque stability
remains unclear.
In the present study, carotid atherosclerotic
plaques samples were obtained from patients undergoing carotid
endarterectomies (CEA). Carotid atherosclerotic plaques were
divided into stable and vulnerable groups. The expression levels of
various types of MMPs, vascular endothelial growth factor (VEGF),
BSP and OPN were compared between the two groups. The current study
provided insight into the development of an improved method of
plaque vulnerability diagnosis, and a possible therapy aimed at
inhibiting plaque formation and stabilizing plaques to prevent
plaque rupture.
Materials and methods
Carotid plaque specimens
Carotid plaques were collected from patients
undergoing CEA in the Department of Neurosurgery of the First
Affiliated Hospital of Soochow University (Suzhou, China). A total
of 64 patients (56 men and 8 women, age 54–82 years) were recruited
between February 2014 and February 2016. All patients underwent
carotid duplex ultrasound, magnetic resonance imaging, computer
tomography perfusion (CTP), computer tomography angiography (CTA)
and/or digital subtraction angiography (DSA) prior to and following
surgery. Plaques were defined as vulnerable if they had any one of
the following characteristics: i) Active inflammation
(monocyte/macrophage infiltration, occasionally with T lymphocyte
infiltration); ii) a thin fibrous cap (thickness <65 um),
accompanied by large lipid core (lipid composition >40%); iii)
endothelial cell loss with platelet aggregation on the surface of
plaques; iv) rupture of the fibrous cap; v) severe vessel stenosis
>90%; or if they had a minimum of two of the following
characteristics: i) Calcified nodules in plaque surface; ii)
intraplaque hemorrhage; iii) dysfunction of endothelial cells in
the plaque; iv) vascular remodeling (25). Based on the aforementioned criteria,
the atherosclerotic carotid plaque samples were divided into 30
stable plaques and 34 vulnerable plaques. Written consent was
obtained from each patient. The experimental protocol was approved
by the Ethics Committee of the First Affiliated Hospital of Soochow
University (approval no. 2011310044).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
The plaque segments were homogenized in liquid
nitrogen. Total RNA was extracted from the homogenates using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). RNA concentration was quantified using a NanoDrop™ 2000
Spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA was
synthesized using the ReverTra Ace® qPCR RT kit (Toyobo
Life Science, Osaka, Japan) according to the manufacturer's
protocol. Primers were synthesized by and purchased from Sangon
Biotech Co., Ltd. (Shanghai, China) (Table I). qPCR analysis was performed using
SYBR® Green Realtime PCR Master mix (Toyobo Life
Science) and an ABI PRISM 7000 sequence detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions were as follows: 95°C for 60 sec, 40 cycles of 95°C for
15 sec and 60°C for 60 sec. Relative mRNA expression was quantified
using the 2−ΔΔCq method (26). GAPDH served as an internal reference
gene. All RT-qPCR analyses were repeated three times.
 | Table I.Sequences of primers used for tissue
sample of human. |
Table I.
Sequences of primers used for tissue
sample of human.
|
| Primer sequence
(5′-3′) |
|---|
|
|
|
|---|
| Gene | Forward | Reverse |
|---|
| MMP-1 |
GAAGAATGATGGGAGGCAAG |
CAGGGTTTCAGCATCTGGTT |
| MMP-2 |
TATGGCTTCTGCCCTGAGAC |
CACACCACATCTTTCCGTCA |
| MMP-3 |
ATCCCGAAGTGGAGGAAAAC |
AGCCTGGAGAATGTGAGTG |
| MMP-7 |
ACCGTGCTGTGTGCTGTGT |
GTCCTGAGCCTGTTCCCACT |
| MMP-9 |
TCTTCCCCTTCACTTTCCTG |
CCCACTTCTTGTCGCTGTC |
| MMP-12 |
CATGAACCGTGAGGATGTTG |
GGCAAAAACCACCAAAATG |
| MMP-13 |
GCAGTCTTTCTTCGGCTTAGAG |
GTATTCACCCACATCAGGAACC |
| MMP-14 |
GCAGAAGTTTTACGGCTTGC |
GCAGAAGTTTTACGGCTTGC |
| MMP-15 |
TTATGGCTACCTGCCTCAGC |
TCCACTCCTTGGTCTCTTCG |
| MMP-16 |
TGATTTACAGGGCATCCAGA |
TCATTTTTCCTTGGGTCAGC |
| MMP-17 |
TGACCACACGAGGCACAT |
CCATCCAGCACTTTCCAGTA |
| MMP-24 |
TGGAGGCAAAAACACATCAC |
TCAAAGGTCAGTGGGGTCA |
| MMP-25 |
GCAGCAACTCTATGGGAAGG |
ATCAGGGATGGGGAAGGAT |
| GAPDH |
AATCCCATCACCATCTTCCA |
AAATGAGCCCCAGCCTTCT |
Protein extraction and western
blotting
The plaque segments were homogenized in liquid
nitrogen and lysed with mammalian protein extraction reagent
supplemented with 1% protease inhibitors (both Beijing ComWin
Biotech, Co., Ltd., Beijing, China) at 4°C for 30 min. Protein
concentration was determined by a BCA assay using the
bicinchonininc acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Protein samples (10 µg) were mixed with an
SDS-PAGE protein loading buffer (Beyotime Institute of
Biotechnology, Haimen, China) and boiled for 10 min. Proteins were
separated on a 12% SDS-PAGE gel and transferred to nitrocellulose
(NC) membranes (EMD Millipore, Billerica, MA, USA). The NC
membranes were then blocked with 5% fat free milk in Tris-buffered
saline with 0.5% Tween-20 (TBST) for 1 h at room temperature. The
membranes were incubated with primary antibodies at 4°C overnight.
The primary antibodies used in the current study were as follows:
Mouse anti-MMP2 (1:1,000; cat. no. Ab80737), rabbit anti-MMP14
(1:1,000; cat. no. Ab51074), mouse anti-MMP25 (1:1,000; cat. no.
GR26383-3), rabbit anti-OPN (1:1,000; cat. no. Ab104302) (all
Abcam, Cambridge UK), mouse anti-VEGF (1:1,000; cat. no. 500-M88;
PeproTech, Rocky Hill, NJ, USA), goat anti-BSP (1:1,000; cat. no
AF4014; R&D Systems, Inc., Minneapolis, MN, USA), rabbit
anti-extracellular regulated kinase (ERK; 1:1,000; cat. no. YT1624)
and rabbit anti-protein kinase C (PKC; 1:1,000; cat. no. YT3752)
(both ImmunoWay Biotechnology Company, Plano, TX, USA) and mouse
anti-GAPDH (1:1,000; cat. no. AG019-1; Beyotime Institute of
Biotechnology). Then, the NC membrane was washed twice with TBST.
The membranes were then incubated with a horseradish
peroxidase-conjugated anti-rabbit secondary antibodies (1:1,000;
cat. no. SA00001-2; ProteinTech Group, Inc., Chicago, IL, USA),
anti-mouse secondary antibodies (1:1,000; A0216) and anti-goat
secondary antibodies (1:1,000; A0181) (both Beyotime Institute of
Biotechnology) for 2 h at room temperature. The protein bands were
developed using an enhanced chemiluminescence kit (Beijing ComWin
Biotech, Co., Ltd.). GAPDH was used as the internal control. The
protein brands were scanned and quantified using ImageJ software
(v1.8.0; National Institutes of Health, Bethesda, MD, USA).
Histology and
immunohistochemistry
Paraffin sections were subjected to
immunohistochemical staining for MMP-2, −14 and −25. The samples
were fixed in 10% formalin and embedded into paraffin at room
temperature until they set. Then, the paraffin section was cut into
4-µm-thick slices. The sections were incubated at 65°C overnight.
Sections were de-paraffinized with xylene for 5 min and re-hydrated
with an alcohol gradient (100, 90 and 70%) for 3 min each time.
Following washing with TBS 3 times, antigen retrieval was performed
and the sections were soaked in 3% H2O2 for
10 min at room temperature. Then, after washing three times the
antigen sites were exposed using a microwave oven with 0.05%
phosphate buffer. The sections were incubated with FBS at 37°C for
30 mins. After washing with PBS 3 times, the sections were
incubated with rabbit and mouse monoclonal antibodies overnight at
4°C. PBS 0.01 M was used to was the tissue sections three times.
The sections were then incubated with horseradish
peroxidase-conjugated mouse anti-rabbit secondary antibodies
(1:500; cat. no. AB11053; BBI Solutions, Cardiff, UK) at 37°C for
30 min. The primary antibodies used in the current study were as
follows: Mouse anti-MMP2 (1:500; cat. no. ab80737), rabbit
anti-MMP14 (1:500; cat. no. ab51074), mouse anti-MMP25 (1:500; cat.
no. GR26383-3) (all Abcam). After washing the following chromogenic
agents were added: 0.05% DAB, 5% H2O2 and
0.05% phosphate buffer. The sections were washed, counterstained
with hematoxylin for 30 sec at room temperature, dehydrated and
protected with coverslips. ImageJ software was used for density
analysis of immunohistochemical results. The results were observed
and captured at magnification, ×200 under an electron microscope.
The gradational distinction was based on positivity cell numbers
and staining intensity. The indicators +, ++ and +++ were used to
indicate the strength of the stain as follows: +, light staining of
plaques as bright yellow; ++, medium staining of plaques as pale
brown; +++, dark staining of plaques as dark brown. Plaque and
tissue sections were also stained with hematoxylin and eosin
(H&E) for 2–3 min at room temperature, for histologic
observation.
Statistical analysis
All statistical analyses were performed with SPSS
20.0 software (IBM Corp, Armonk, NY, USA). Data were analyzed by
paired t-tests or, for immunohistochemical results, Chi-squared
tests. Values are presented as mean ± standard deviation. P<0.05
was considered to indicate a statistically significant
difference.
Results
Patient characteristics and clinical
data
As determined by ultrasonography and H&E
staining, all the carotid plaques were divided into 30 stable
plaques and 34 vulnerable plaques. As presented in Fig. 1A, stable plaques exhibited uniform
echoes and smooth surfaces. Conversely, vulnerable plaques
exhibited uneven echoes and irregular shapes, and color Doppler
blood signals were occasionally exhibited (Fig. 1B). The H&E staining of the stable
plaques revealed the smooth and thick fibrous cap (Fig. 1C). In contrast, the majority of
vulnerable plaques contained discontinuous and thin fibrous caps
with thrombi, neovascularization and lipid cores (Fig. 1D). Table
I presents the demographics, risk factors for ischemic stroke,
degrees of stenosis, diseases and clinical symptoms of the patients
involved in the present study. As demonstrated in Table I, the development of vulnerable
plaques was associated with sex. Males were significantly more
likely to develop vulnerable carotid plaques compared with females.
In addition, patients with vulnerable plaques had a statistically
higher number of transient ischemic attacks (TIA; Table II). No significant differences were
identified in the other symptoms and stroke risk factors between
the two groups.
| Table II.Clinical index of the 64 patients
recruited in this study. |
Table II.
Clinical index of the 64 patients
recruited in this study.
| Characteristic | Stable (n=30) | Vulnerable
(n=34) | P-value |
|---|
| Age mean
(years) | 67 | 69 | NS |
| Sex
(male/female) | 23/7 | 33/1 |
<0.05a |
| Amaurosis
fugax | 3
(10%) | 5
(15%) | NS |
| Transient ischemic
attack | 5
(17%) | 13 (38%) | <0.05 |
| Stroke | 4
(13%) | 7
(20%) | NS |
| Hypertension | 6
(20%) | 9
(26%) | NS |
| Diabetes | 3 (6%) | 4
(12%) | NS |
| Hyperlipidemia | 7
(23%) | 8
(24%) | NS |
| Smoking habit | 12 (40%) | 15 (44%) | NS |
| Statins | 16 (53%) | 14 (44%) | NS |
| Degree of stenosis
(medium/slight) | 4/26 | 7/27 | NSa |
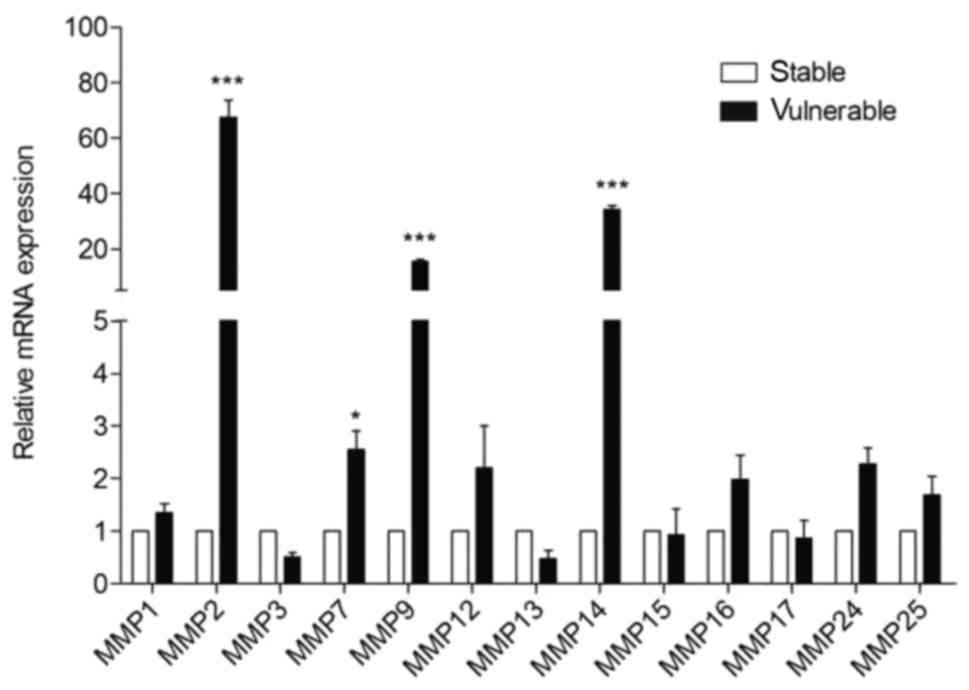
mRNA levels of MMP-2, −7, −9 and −14
are upregulated in vulnerable plaques
MMPs are metalloproteases important in the
destabilization of plaques (27). To
understand which metalloproteases are determinants for the onset of
vulnerable plaques, the mRNA levels of various MMPs were analyzed
in the plaque segments. The mRNA level of MMP-1, −2, −3, −7, −9,
−12, −13, −14, −15, −16, −17, −24 and −25 were compared between the
stable and vulnerable plaque segments (Fig. 2). The expression levels of MMP-2, −9,
−14 were significantly higher (>20-fold) in the vulnerable
plaques compared with the stable plaques; MMP-7 expression levels
were also significantly raised (>2-fold) in vulnerable plaques
compared with stable plaques. No significant differences were
identified in the mRNA levels of other MMPs between the two plaque
types.
MMP-2 and MMP-14 protein levels are
increased in homogenized vulnerable plaques
MMP-7 and MMP-9 have been extensively studied prior
to the current study (28–30), therefore MMP-2 and MMP-14 were
selected for quantification by western blotting; MMP-2 and MMP-14
also demonstrated the greatest expression fold change in Fig. 2. MMP-25, whose expression was not
altered between the two types of plaques, was selected as a
negative control. Consistent with the gene expression, the protein
levels of MMP-2 and MMP-14 were significantly upregulated
(>2-fold) in vulnerable plaques compared with those in stable
plaques (Fig. 3A). Consistent with
the mRNA level, no significant difference was identified in the
protein levels of MMP-25 between the two groups.
 | Figure 3.Protein level of MMPs, associated
factors and signaling pathway proteins altered in vulnerable
plaques. Western blot analyses and quantification of (A) MMP-2,
MMP-14, MMP-25, VEGF, (B) BSP, OPN, ERK and PKC protein levels in
stable and vulnerable plaques. Data are presented the mean ±
standard deviation. *P<0.05, **P<0.01 and ***P<0.001 vs.
the stable plaques group. MMP, matrix metalloproteinase; VEGF,
vascular endothelial growth factor; BSP, bone sialoprotein 2; OPN,
osteopontin; ERK, mitogen-activated protein kinase 3; PKC, protein
kinase C. |
Angiogenesis and calcification factors
are altered in vulnerable plaques
To further investigate the biochemical
characteristics of different types of plaques, the protein levels
of vascularization marker VEGF and calcification markers BSP and
OPN were analyzed. VEGF expression was significantly upregulated in
vulnerable plaques compared with the stable group (Fig. 3A). The protein expression of BSP was
significantly increased while OPN protein levels were significantly
decreased in vulnerable plaques compared with stable plaques
(Fig. 3B).
To gain more insight into the signaling pathway that
is activated and may mediate the pathogenesis of plaque
vulnerability, the steady state protein level of key mediators of
various signaling pathways was also analyzed. No significant
differences were identified in proto-oncogene c-Fos, RAC-alpha
serine/threonine-protein kinase, runt-related transcription factor
2, protein Wnt-16, mothers against decapentaplegic homolog (SMAD)1,
SMAD2,3 proteins between the stable and vulnerable groups of
carotid plaques (data not shown). However, the steady state protein
levels of ERK and PKC were significantly increased in vulnerable
plaques compared with stable plaques (Fig. 3B).
Protein levels of MMP-2 and MMP-14 are
increased in vulnerable plaque tissue sections
The protein levels of MMP-2 and MMP-14 were
upregulated in vulnerable plaques compared with stable plaques. To
localize MMP-2 and MMP-14 expression within the two groups of
carotid plaque, serial tissue sections were subjected to
immunostaining (Fig. 4). H&E
staining revealed that macrophages and foam cells were primarily
distributed at the location of the plaque rupture and were more
widely distributed in the vulnerable plaque group than in the
stable plaque group. The two groups of carotid plaques were
positively stained with MMP-2 and MMP-14 at different intensities.
Staining for MMP-2 and MMP-14 was markedly more intense in
vulnerable plaques compared with stable plaques. MMP-2 and MMP-14
staining was positive in the atherosclerotic lesions, and markedly
increased in the breakdown region of the fibrous cap. Consistent
with the protein level, density analysis revealed that the
immunohistochemical staining of MMP-2 (Table III) and MMP-14 (Table IV) was significantly more intense in
samples from vulnerable plaques compared with stable plaques. It
was observed that staining for MMP-25 was not significantly
different between the vulnerable and stable plaques (data not
shown).
| Table III.Statistics of immunohistochemical
matrix metalloproteinase 2 staining in stable and vulnerable
plaques. |
Table III.
Statistics of immunohistochemical
matrix metalloproteinase 2 staining in stable and vulnerable
plaques.
| Group | + | ++ | +++ | Total |
|---|
| Stable | 16 | 10 | 4 | 30 |
|
Vulnerablea | 3 | 12 | 19 | 34 |
| Total | 19 | 22 | 23 | 64 |
| Table IV.Statistics of immunohistochemical
matrix metalloproteinase 14 staining in stable and vulnerable
plaques. |
Table IV.
Statistics of immunohistochemical
matrix metalloproteinase 14 staining in stable and vulnerable
plaques.
| Group | + | ++ | +++ | Total |
|---|
| Stable | 17 | 11 | 2 | 30 |
|
Vulnerablea | 5 | 13 | 16 | 34 |
| Total | 22 | 24 | 18 | 64 |
Discussion
Atherosclerosis is the critical risk factor
associated with the onset of stroke. Carotid stenosis is a serious
long-term outcome of atherosclerosis (31,32).
Carotid atherosclerotic plaques, particularly vulnerable carotid
plaques were closely associated with ischemic stroke (33). In the current study, 64 patients were
recruited and carotid plaques were collected following CEA. The
collected carotid plaques were divided into the stable and
vulnerable groups. Patients with vulnerable plaques had
significantly higher numbers of TIA. According to previous studies,
these patients exhibit a higher risk of acute cerebrovascular
events caused by the rupture of vulnerable plaques (34–36).
An increasing number of studies have suggested that
MMPs are the foremost factors that degrade the ECM and cause the
rupture of vulnerable plaques (37,38).
MMPs are grouped into five types based on their structure and
substrate specificity; these include collagenases (MMP-1, −8, −13
and −18), gelatinases (MMP-2 and −9), stromelysins (MMP-3, −7, −10,
−11 and −12), membrane-type MMPs (MMP-14, −15, −16, −17, −24 and
−25) and the other MMPs (39). In
the current study, the expression levels of various MMPs in human
carotid plaques were investigated; it was demonstrated that the
expression levels of MMP-2, −7, −9 and −14 were elevated in
vulnerable plaques, particularly MMP-2 and −14. Additionally,
immunohistochemistry demonstrated that MMP-2 and −14 were
predominantly distributed in the ruptured region of vulnerable
plaques, which was the same area where VSMCs and macrophage
aggregated (40). Studies have
confirmed that the overexpression of MMP-2 and −14 serves a
critical role in the cell migration and growth of endothelial
cells, and the formation of new blood vessels by adjusting the ECM
environment (41,42).
The activation of MMP-2 was associated with the
increased expression of MMP-14 (41). MMP-14 not only acts as a protease,
but also a signaling molecule, which affects the pathological and
physiological functions of cells (42). As is well known, the ECM is important
for remodeling the fibrous cap (43). Vulnerable plaques are morphologically
characterized as possessing a thin fibrous cap (44). In the present study, elevated
expression of MMP-2 and −14 was observed, which may lead to ECM
degradation, resulting in further thinning of the shoulders of the
fibrous cap and the rupture of plaques. Previous studies have
suggested that the expression of MMP-25 is low in atherosclerosis
(45,46). Notably, no significant difference in
MMP-25 protein level was identified between stable and vulnerable
plaques in the present study, suggesting that MMP-25 did not
contribute to the plaque vulnerability and may have no role in the
rupture of vulnerable plaques. The data from the current study
indicated that MMP-2 and −14 may be valuable clinical biomarkers
for plaque vulnerability.
VEGF serves a decisive role in promoting instability
by inducing angiogenesis in plaques (47). The current study verified that the
expression of VEGF was significantly higher in vulnerable plaques.
Angiogenesis is a critical factor in the process of
atherosclerosis, which can induce intraplaque hemorrhage and
rupture. Plaque calcification gradually forms during
atherosclerosis (48). Various
studies suggested that calcification may alter the stability of
atherosclerotic plaques (24,49).
During calcification, VSMCs synthesize several osteogenic factors,
including BSP (50). Fedarko et
al (51) revealed that BSP
specifically binds proMMP-2 and active MMP-2, while OPN binds
proMMP-3 and active MMP-3. It has been demonstrated that BSP
exhibits hydroxyapatite nucleation activity and may serve important
roles in the initiation of and calcification during atherosclerosis
(52,53). OPN is a potent inhibitor of
mineralization (54). OPN prevents
ectopic calcium deposition and is a strong, inducible inhibitor of
vascular calcification (55). The
current study revealed that the overexpression of BSP and
attenuation of OPN may be associated with plaque vulnerability.
A number of studies investigating the association
between MMPs and vascular biology have demonstrated that a number
of factors leading to angiogenesis and vascular
remodeling-associated diseases regulate MMP expression and
activation, including hemodynamics, oxidative stress, inflammation,
hormonal factors and hypoxia (56,57).
Studies have revealed that low fluid shear stress-induced MMP
expression involves the integrin, p38 mitogen-activated protein
kinase 1 or ERK1/2-nuclear factor (NF)-κB signaling pathways
(58,59). Studies have indicated that OPN may
promote the expression of MMP-2 through the ERK signaling pathways
(56,60). In the present study, various
signaling pathways were investigated by western blotting and it was
demonstrated that ERK and PKC were significantly higher in
vulnerable plaques, indicating that they may be involved the
process of plaque rupture. It was hypothesized that activation of
ERK and PKC may also increase MMP-2 and −14 expression.
Simultaneously, ERK and PKC were activated to promote BSP secretion
and decrease OPN expression, which promotes the upregulation of
MMP-2 and −14, and increase the likelihood of shoulder rupture of
atherosclerotic plaques.
In summary, the current study demonstrated that
MMP-2 and −14 were elevated in vulnerable plaques and may serve a
significant role in rupture of carotid plaques. The differential
regulation of factors, including VEGF and BSP, were also associated
with the plaque vulnerability. More studies are required to
demonstrate the association of the aforementioned factors with
plaque rupture events and the accurate prediction of vulnerable
plaques. Mechanistic studies investigating how plaque rupture is
regulated should be performed for the development of possible
therapies that stabilize vulnerable plaques and reduce the
occurrence of ischemic stroke.
Acknowledgements
The authors would like to thank Professor Lan Xu
(Soochow University, Suzhou, China) for her assistance and
advice.
Funding
This work was supported by the National Natural
Science Foundation of China (grant no. 31401173) and the Natural
Science Foundation of Jiangsu Province (grant no. BK20140329). This
work was also supported by the People's Livelihood Technology
Demonstration Project of Suzhou (grant no. SS201714) and the
Province Care Health Care Research Project of Jiangsu (grant no.
BJ17010).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZYG contributed to the acquisition and analysis of
the data, performed the basic experiments and was involved in the
drafting of the manuscript. PJH contributed to the conception and
design of the study and gave final approval of the version to be
published. LX contributed to the conception and design of the data
analysis, assisted with the experiments and was involved in
revising the manuscript critically for intellectual content. BZ and
YHY assessed the properties of the plaques using vascular
ultrasound and assisted with the collection of specimens. SSG and
JJL assisted with the experiments. All authors read and approved
the final version of the manuscript.
Ethics approval and consent to
participate
Written consent was obtained from each patient. The
experimental protocol was approved by the ethics committee of the
First Affiliated Hospital of Soochow University (approval no.
2011310044).
Patient consent for publication
The patients have provided written informed consent
for the publication of any associated data and accompanying
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu XS, Zhao HL, Cao Y, Lu Q and Xu JR:
Comparison of carotid atherosclerotic plaque characteristics by
high-resolution black-blood MR imaging between patients with
first-time and recurrent acute ischemic stroke. AJNR Am J
Neuroradiol. 33:1257–1261. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosamond W, Flegal K, Friday G, Furie K,
Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, et al: Heart
disease and stroke statistics-2007 update: A report from the
American Heart Association Statistics Committee and Stroke
Statistics Subcommittee. Circulation. 115:e69–e171. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kuk M, Wannarong T, Beletsky V, Parraga G,
Fenster A and Spence JD: Volume of carotid artery ulceration as a
predictor of cardiovascular events. Stroke. 45:1437–1441. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jin G: The relationship between serum
CXCL16 level and carotid vulnerable plaque in patients with
ischemic stroke. Eur Rev Med Pharmacol Sci. 21:3911–3915.
2017.PubMed/NCBI
|
|
5
|
Chatzizisis YS, Antoniadis AP, Wentzel JJ
and Giannoglou GD: Vulnerable plaque: The biomechanics of matter.
Atherosclerosis. 236:351–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Badimon L and Vilahur G: Thrombosis
formation on atherosclerotic lesions and plaque rupture. J Intern
Med. 276:618–632. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yonetsu T and Jang IK: Advances in
Intravascular Imaging: New insights into the vulnerable plaque from
imaging studies. Korean Circ J. 48:1–15. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Virmani R, Burke AP, Farb A and Kolodgie
FD: Pathology of the vulnerable plaque. J Am Coll Cardiol. 47 8
Suppl:C13–C18. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Souilhol C, Harmsen MC, Evans PC and
Krenning G: Endothelial-mesenchymal transition in atherosclerosis.
Cardiovasc Res. 114:565–577. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Heeneman S, Cleutjens JP, Faber BC,
Creemers EE, van Suylen RJ, Lutgens E, Cleutjens KB and Daemen MJ:
The dynamic extracellular matrix: Intervention strategies during
heart failure and atherosclerosis. J Pathol. 200:516–525. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Galis ZS, Sukhova GK, Lark MW and Libby P:
Increased expression of matrix metalloproteinases and matrix
degrading activity in vulnerable regions of human atherosclerotic
plaques. J Clin Invest. 94:2493–2503. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jones CB, Sane DC and Herrington DM:
Matrix metalloproteinases: A review of their structure and role in
acute coronary syndrome. Cardiovasc Res. 59:812–823. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singh T, Adekoya OA and Jayaram B:
Understanding the binding of inhibitors of matrix
metalloproteinases by molecular docking, quantum mechanical
calculations, molecular dynamics simulations, and a MMGBSA/MMBappl
study. Mol Biosyst. 11:1041–1051. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nagase H and Woessner JF Jr: Matrix
metalloproteinases. J Biol Chem. 274:21491–21494. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Graham CA, Chan RW, Chan DY, Chan CP, Wong
LK and Rainer TH: Matrix metalloproteinase 9 mRNA: An early
prognostic marker for patients with acute stroke. Clin Biochem.
45:352–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Heo W, Lee YS, Son CH, Yang K, Park YS and
Bae J: Radiation-induced matrix metalloproteinases limit natural
killer cell-mediated anticancer immunity in NCI-H23 lung cancer
cells. Mol Med Rep. 11:1800–1806. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han Y, Mao X, Wang L, Liu J, Wang D, Cheng
H and Miao G: increased levels of soluble cluster of
differentiation 40 ligand, matrix metalloproteinase 9, and matrix
metalloproteinase 2 are associated with carotid plaque
vulnerability in patients with ischemic cerebrovascular disease.
World Neurosurg. 105:709–713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Virmani R, Kolodgie FD, Burke AP, Finn AV,
Gold HK, Tulenko TN, Wrenn SP and Narula J: Atherosclerotic plaque
progression and vulnerability to rupture: Angiogenesis as a source
of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol.
25:2054–2061. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Foley CJ, Fanjul-Fernández M, Bohm A,
Nguyen N, Agarwal A, Austin K, Koukos G, Covic L, López-Otín C and
Kuliopulos A: Matrix metalloprotease 1a deficiency suppresses tumor
growth and angiogenesis. Oncogene. 33:2264–2272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Doherty TM, Asotra K, Fitzpatrick LA, Qiao
JH, Wilkin DJ, Detrano RC, Dunstan CR, Shah PK and Rajavashisth TB:
Calcification in atherosclerosis: Bone biology and chronic
inflammation at the arterial crossroads. Proc Natl Acad Sci USA.
100:11201–11206. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wahlgren CM, Zheng W, Shaalan W, Tang J
and Bassiouny HS: Human carotid plaque calcification and
vulnerability. Relationship between degree of plaque calcification,
fibrous cap inflammatory gene expression and symptomatology.
Cerebrovasc Dis. 27:193–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Charo IF and Ransohoff RM: The many roles
of chemokines and chemokine receptors in inflammation. N Engl J
Med. 354:610–621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Charo IF and Taubman MB: Chemokines in the
pathogenesis of vascular disease. Circ Res. 95:858–866. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kruger TE, Miller AH, Godwin AK and Wang
J: Bone sialoprotein and osteopontin in bone metastasis of
osteotropic cancers. Crit Rev Oncol Hematol. 89:330–341. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Naghavi M, Libby P, Falk E, Casscells SW,
Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P,
Pasterkamp G, et al: From vulnerable plaque to vulnerable patient:
A call for new definitions and risk assessment strategies: Part I.
Circulation. 108:1664–1672. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Müller A, Krämer SD, Meletta R, Beck K,
Selivanova SV, Rancic Z, Kaufmann PA, Vos B, Meding J, Stellfeld T,
et al: Gene expression levels of matrix metalloproteinases in human
atherosclerotic plaques and evaluation of radiolabeled inhibitors
as imaging agents for plaque vulnerability. Nucl Med Biol.
41:562–569. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hakimzadeh N, Pinas VA, Molenaar G, de
Waard V, Lutgens E, van Eck-Smit BLF, de Bruin K, Piek JJ, Eersels
JLH, Booij J, et al: Novel molecular imaging ligands targeting
matrix metalloproteinases 2 and 9 for imaging of unstable
atherosclerotic plaques. PLoS One. 12:e01877672017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Razuvaev A, Ekstrand J, Folkersen L,
Agardh H, Markus D, Swedenborg J, Hansson GK, Gabrielsen A,
Paulsson-Berne G, Roy J and Hedin U: Correlations between clinical
variables and gene-expression profiles in carotid plaque
instability. Eur J Vasc Endovasc Surg. 42:722–730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ragino IuI, Cherniavskiĭ AM, Polonskaia
IaV, Volkov AM, Semaeva EV, Tsymbal SIu and Voevoda MI: Changes in
proinflammatory cytokine and destructive metalloproteinase levels
during formation of unstable atherosclerotic plaque. Kardiologiia.
49:43–49. 2009.(In Russian). PubMed/NCBI
|
|
31
|
Andrews JPM, Fayad ZA and Dweck MR: New
methods to image unstable atherosclerotic plaques. Atherosclerosis.
272:118–128. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lapenna D, Ciofani G, Pierdomenico SD,
Giamberardino MA, Ucchino S and Davì G: Association of serum
bilirubin with oxidant damage of human atherosclerotic plaques and
the severity of atherosclerosis. Clin Exp Med. 18:119–124. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pelisek J, Eckstein HH and Zernecke A:
Pathophysiological mechanisms of carotid plaque vulnerability:
Impact on ischemic stroke. Arch Immunol Ther Exp (Warsz).
60:431–442. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sakakura K, Nakano M, Otsuka F, Ladich E,
Kolodgie FD and Virmani R: Pathophysiology of atherosclerosis
plaque progression. Heart Lung Circ. 22:399–411. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rai V and Agrawal DK: The role of damage-
and pathogen-associated molecular patterns in inflammation-mediated
vulnerability of atherosclerotic plaques. Can J Physiol Pharmacol.
95:1245–1253. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jain M, Wu B, Pisapia D, Salvatore S,
Mukherjee S and Narula N: A component-by-component characterisation
of high-risk atherosclerotic plaques by multiphoton microscopic
imaging. J Microsc. 268:39–44. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ulrich V, Rotllan N, Araldi E, Luciano A,
Skroblin P, Abonnenc M, Perrotta P, Yin X, Bauer A, Leslie KL, et
al: Chronic miR-29 antagonism promotes favorable plaque remodeling
in atherosclerotic mice. EMBO Mol Med. 8:643–653. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Eilenberg W, Stojkovic S, Kaider A,
Kozakowski N, Domenig CM, Burghuber C, Nanobachvili J, Huber K,
Klinger M, Neumayer C, et al: NGAL and MMP-9/NGAL as biomarkers of
plaque vulnerability and targets of statins in patients with
carotid atherosclerosis. Clin Chem Lab Med. 56:147–156. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Visse R and Nagase H: Matrix
metalloproteinases and tissue inhibitors of metalloproteinases:
Structure, function, and biochemistry. Circ Res. 92:827–839. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Prebble H, Cross S, Marks E, Healy J,
Searle E, Aamir R, Butler A, Roake J, Hock B, Anderson N and Gieseg
SP: Induced macrophage activation in live excised atherosclerotic
plaque. Immunobiology. 223:526–535. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen Q, Jin M, Yang F, Zhu J, Xiao Q and
Zhang L: Matrix metalloproteinases: Inflammatory regulators of cell
behaviors in vascular formation and remodeling. Mediators Inflamm.
2013:9283152013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ohkawara H, Ikeda K, Ogawa K and Takeishi
Y: Membrane Type 1-matrix metalloproteinase (Mt1-Mmp) identified as
a multifunctional regulator of vascular responses. Fukushima J Med
Sci. 61:91–100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Korol RM, Canham PB, Liu L, Viswanathan K,
Ferguson GG, Hammond RR, Finlay HM, Baker HV, Lopez C and Lucas AR:
Detection of altered extracellular matrix in surface layers of
unstable carotid plaque: An optical spectroscopy, birefringence and
microarray genetic analysis. Photochem Photobiol. 87:1164–1172.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rohwedder I, Montanez E, Beckmann K,
Bengtsson E, Dunér P, Nilsson J, Soehnlein O and Fässler R: Plasma
fibronectin deficiency impedes atherosclerosis progression and
fibrous cap formation. EMBO Mol Med. 4:564–576. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Newby AC: Metalloproteinases promote
plaque rupture and myocardial infarction: A persuasive concept
waiting for clinical translation. Matrix Biol. 44–46:157–166. 2015.
View Article : Google Scholar
|
|
46
|
Starr AE, Bellac CL, Dufour A, Goebeler V
and Overall CM: Biochemical characterization and N-terminomics
analysis of leukolysin, the membrane-type 6 matrix metalloprotease
(MMP25): Chemokine and vimentin cleavages enhance cell migration
and macrophage phagocytic activities. J Biol Chem. 287:13382–13395.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Michel JB, Martin-Ventura JL, Nicoletti A
and Ho-Tin-Noé B: Pathology of human plaque vulnerability:
Mechanisms and consequences of intraplaque haemorrhages.
Atherosclerosis. 234:311–319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gopalakrishnan M, Silva-Palacios F,
Taytawat P, Pant R and Klein L: Role of inflammatory mediators in
the pathogenesis of plaque rupture. J Invasive Cardiol. 26:484–492.
2014.PubMed/NCBI
|
|
49
|
Golledge J, McCann M, Mangan S, Lam A and
Karan M: Osteoprotegerin and osteopontin are expressed at high
concentrations within symptomatic carotid atherosclerosis. Stroke.
35:1636–1641. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Farrokhi E, Samani KG and Chaleshtori MH:
Oxidized low-density lipoprotein increases bone sialoprotein
expression in vascular smooth muscle cells via runt-related
transcription factor 2. Am J Med Sci. 349:240–243. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fedarko NS, Jain A, Karadag A and Fisher
LW: Three small integrin binding ligand N-linked glycoproteins
(SIBLINGs) bind and activate specific matrix metalloproteinases.
FASEB J. 18:734–736. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Harris NL, Rattray KR, Tye CE, Underhill
TM, Somerman MJ, D'Errico JA, Chambers AF, Hunter GK and Goldberg
HA: Functional analysis of bone sialoprotein: Identification of the
hydroxyapatite-nucleating and cell-binding domains by recombinant
peptide expression and site-directed mutagenesis. Bone. 27:795–802.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yang Y, Cui Q and Sahai N: How does bone
sialoprotein promote the nucleation of hydroxyapatite? A molecular
dynamics study using model peptides of different conformations.
Langmuir. 26:9848–9859. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Golledge J, McCann M, Mangan S, Lam A and
Karan M: Osteoprotegerin and osteopontin are expressed at high
concentrations within symptomatic carotid atherosclerosis. Stroke.
35:1636–1641. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Higgins CL, Isbilir S, Basto P, Chen IY,
Vaduganathan M, Vaduganathan P, Reardon MJ, Lawrie G, Peterson L
and Morrisett JD: Distribution of alkaline phosphatase,
osteopontin, RANK ligand and osteoprotegerin in calcified human
carotid atheroma. Protein J. 34:315–328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Newby AC: Proteinases and plaque rupture:
Unblocking the road to translation. Curr Opin Lipidol. 25:358–366.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Newby AC: Newby: Dual role of matrix
metalloproteinases (matrixins) in intimal thickening and
atherosclerotic plaque rupture. Physiol Rev. 85:1–31. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Steitz SA, Speer MY, McKee MD, Liaw L,
Almeida M, Yang H and Giachelli CM: Osteopontin inhibits mineral
deposition and promotes regression of ectopic calcification. Am J
Pathol. 161:2035–2046. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gravallese EM: Osteopontin: A bridge
between bone and the immune system. J Clin Invest. 112:147–149.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xu ST, Zou FZ, Cai LN and Xu WL: The
downregulation of OPN inhibits proliferation and migration and
regulate activation of Erk1/2 in ECA-109 cells. Int J Clin Exp Med.
8:5361–5369. 2015.PubMed/NCBI
|