Introduction
Vascular dementia (VD) is one of the most prevalent
forms of dementia, which progressively affects cognitive abilities
due to reduced blood flow in the brain (1–4). In
general, VD patients often exhibit some cognitive disorder, such as
forgetfulness, depression, anxiety or disorientation (5). Unfortunately, the molecular mechanism
of VD remains unidentified at present. Notably, it has been
accepted that ischemic stroke is an important risk factor of VD,
leading to increased mortality and morbidity (6–9).
Ischemic stroke can generate blood-brain barrier (BBB) damage and
brain vasogenic edema. The process includes oxidative stress,
inflammation, apoptosis and excitotoxicity (10–12).
Subsequent cognition impairment caused by BBB damage induced by
stroke has been observed (13).
Therefore, the suppression of BBB damage may help to prevent
ischemic stroke and post-stroke dementia.
The BBB is the major site of blood-central nervous
system exchange at the level of the brain microvessel endothelium
(14,15). Brain microvascular endothelial cells
(BMECs), the primary structures responsible for BBB permeability,
are connected by tight junctions (TJ) and control the ionic
equilibrium in brain (16,17). Increasing evidence has supported the
idea that disorders of some TJ-associated proteins promote the
dysfunction of the BBB in many central nervous system diseases
(18,19). Loss of BBB integrity is one of the
key events in ischemic stroke (20).
Unfortunately, the exact molecular mechanism underlying the BBB
damage associated with stroke has not been elucidated.
The Eph receptor-ligand family represents the
largest family of tyrosine kinases; the interaction between Eph
receptors and their ligands is pivotal to physiological functioning
by regulating cell-cell contact (21). Increasing evidence has shown that the
Eph-Ephrin system is involved in inflammatory processes (22). Interestingly, it has been
demonstrated that EphrinA1/EphA4 signaling is pivotal to the
adhesion between monocytes and endothelial cells in vitro,
thus EphrinA1/EphA4 signaling may play a key role in inflammatory
stimuli (23). Moreover,
inflammatory processes participate in the pathology of ischemic
stroke, contributing to the damage to the BBB (24,25).
Therefore, we hypothesize that the activation of EphrinA1/EphA4
signaling promotes stroke-induced BBB damage and brain injury.
In our research, we detected the effect of
EphrinA1/EphA4 signaling on BBB damage following cerebral
ischemia-reperfusion (I/R) in mice. We first demonstrated that
EphrinA1/EphA4 signaling could enhance ischemic brain injury via
the Rho/ROCK pathway in vivo, which was associated with
increased BBB permeability, as indicated by down-regulation of
TJ-associated proteins and enhanced inflammatory cell
infiltration.
Materials and methods
Reagents
Primary antibodies targeting EphrinA1, EphA4,
Phospho-ROCK, ROCK, CD68, ZO-1 and Claudin-5 were obtained from
Abcam (Cambridge, MA, USA). Recombinant Mouse EphrinA1 protein and
EphA4-Fc were purchased from R&D Systems, Inc. (Minneapolis,
MN, USA). Primary antibodies targeting caspase3, Phospho-AKT and
AKT were purchased from Beyotime Institute of Biotechnology
(Shanghai, China). The primary antibody targeting β-actin was
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Cell culture
Human brain microvascular endothelial cells
(HBMECs), which retain key features of cerebral endothelial
function were obtained from Cell Systems Corporation (cat. no.
ACBRI376; Kirkland, WA, USA) and used for the in vitro
assay. The cells were maintained in DMEM supplemented with 10%
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA), 100 IU/ml penicillin, 100 IU/ml streptomycin and 5%
CO2 in a humidified incubator at 37°C.
In vivo stroke model
In this study, the mouse model of stroke was induced
via the method of middle cerebral artery occlusion (MCAO), as
previously described (26). All
experiments were approved by the Medical Ethics Committee of the
PLA 102nd Hospital for Animal Care and Use. A total of 40 male mice
(20–24 g) were used to detect the effect of the EphrinA1/EphA4
signaling pathway in I/R injury. These mice were divided into four
groups: Sham (n=10), MCAO (n=10), MCAO+EphrinA1 (n=10) and
MCAO+EphrinA1+EphA4-Fc (n=10). In the MCAO groups, temporary left
MCAO was performed for 60 min; the mice were anesthetized using 5%
chloral hydrate (400 mg/kg), and then a midline ventral neck
incision was made. A 6-0 nylon monofilament (0.20–0.22 mm tip) was
used to obstruct the artery lumen, followed by reperfusion for 72 h
in the MCAO groups. In the sham group, arteries were visualized but
not disturbed. The mice in the 72 h I/R groups were assigned to
receive a caudal vein infusion of EphrinA1 (2 mg/kg/d) or EphA4-Fc
(1 mg/kg/d) at 1 h after reperfusion. In addition, a total of 40
male mice (20–24 g) were used to detect the effect of the Rho/Rock
signaling pathway in EphrinA1/EphA4 system-mediated I/R injury;
these mice were divided into five groups: Sham (n=8), MCAO (n=8),
MCAO+EphrinA1 (n=8), MCAO+EphrinA1+C3 transferase (n=8) and
MCAO+EphrinA1+Y-27,632 (n=8). Rho inhibitor (C3 transferase) (4
mg/kg/d) or ROCK inhibitor (Y-27,632) (5 mg/kg/d) was infused via
the caudal vein, with the first administration at 30 min prior to
EphrinA1 after reperfusion. All samples in this study were
collected at 72 h after reperfusion.
Western blotting
The harvested cortical tissues at 24, 48 and 72 h
after MCAO were homogenized, then lysed in RIPA buffer (Beyotime
Institute of Biotechnology, Shanghai, China) containing 1% protease
inhibitor (Thermo Fisher Scientific, Inc.). After collecting the
protein, the protein levels were quantified using a BCA protein
assay kit (Thermo Fisher Scientific, Inc.). Subsequently, 50 µg
proteins were loaded, electrophoresed and transferred onto
polyvinylidene difluoride membranes, which were blocked for 1.5 h
using 5% milk in TBST. The membranes were incubated with primary
antibodies overnight at 4°C. After three washes, IgG-HRP secondary
antibody (1:5,000 dilution) (Zhonshan Biology Company, Beijing,
China) incubation was conducted for 1 h at 37°C. Finally, the
membranes were stained with DAB mixed liquid (Bio-Rad Laboratories,
Inc., Hercules, CA, USA), and scanned using a gel imaging
system.
Glucose deprivation
Oxygen-glucose deprivation/reperfusion (OGD/R) was
performed to simulate ischemic conditions in vitro, as
previously described (27). The
HBMEC cultures were subjected to OGD/R conditions by replacing the
normal medium with glucose-free Locke's solution, and were
incubated under 94% N2/5% CO2/1%
O2 at 37°C for 6 h. The cells were then reperfused by
placing them in fresh normal DMEM under normal atmospheric
conditions for 12 or 24 h.
Histological analysis
Brains were collected from all groups and fixed for
histological analysis. The whole brains were extracted and treated
with formalin for 24 h. After dehydration, each brain was embedded
in paraffin and cut into 8-µm sections of tissue using a histotome.
After rehydration, the brain tissue sections were permeabilized
with 0.3% Triton-X and blocked in 5% BSA for 1 h at 37°C, then
incubated with EphrinA1, EphA4 and CD68 antibodies overnight at
4°C. Following primary incubation, the sections were incubated with
HRP-conjugated secondary antibodies for 1 h at 37°C. The
chromogenic reaction was performed using DAB mixed liquid. After
dehydration and treatment with Permount™ mounting medium,
micrographs were captured with a light microscope.
Measurement of brain edema
The wet-dry method was used to measure the degree of
brain edema following I/R injury. The left and right sides of the
brains were divided, and the wet weights of the hemispheres were
recorded immediately. Subsequently, the hemispheres were dried in
the oven for 3 days at 100°C. Brain edema was detected by
calculating the water content based on the following formula: Brain
water content (%)=(1-dry weight/wet weight) ×100%.
Neurological scores
Brain function following I/R injury was examined
using a neurological deficit scoring scale. The scoring system was
defined as follows: 0=no deficit, 1=failure to fully extend the
contralateral forelimb, 2=circling to the contralateral side,
3=leaning to the contralateral side at rest and 4=no spontaneous
walking. Each mouse was scored three times in a blinded manner.
Rho activation assay
Based on the instructions of the Rho activation
measurement kit, HBMEC lysates were incubated with rhotekin
Rho-binding peptide immobilized on agarose. Activated GTP-Rho bound
to rhotekin agarose was detected by anti-Rho antibody (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA).
Statistical analysis
All results are expressed as the mean ± SEM. SPSS
17.0 software (SPSS, Inc., Chicago, IL, USA) was used for analysis.
One-way analysis of variance (ANOVA) was used to examine the
differences in the data of each group, followed by the LSD post hoc
test. Neurological deficit scores were analyzed using a
non-parametric Kruskal-Wallis test. The differences were considered
significant at P<0.05.
Results
Upregulation of EphrinA1 and EphA4
following cerebral I/R in vivo and in vitro
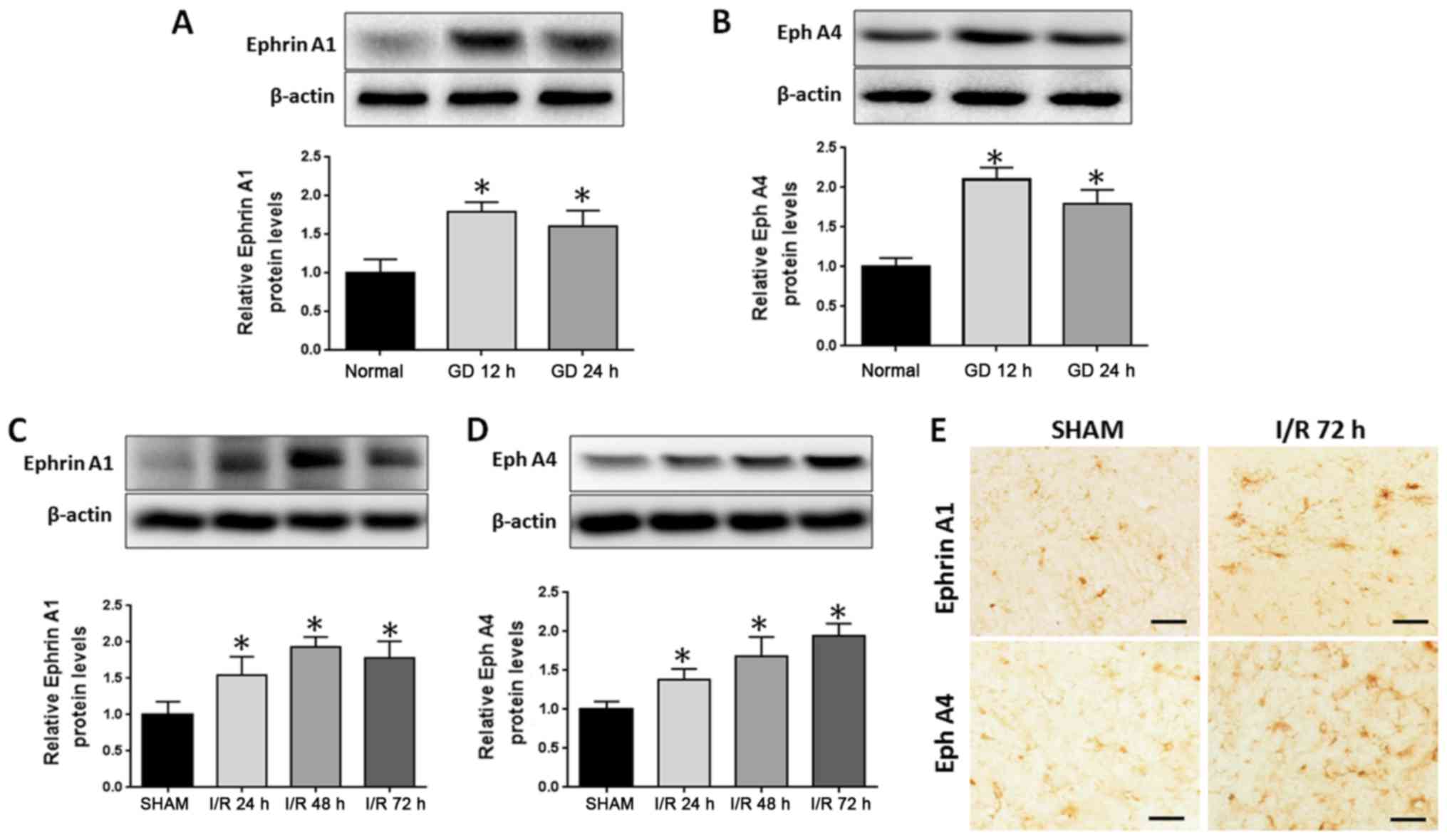
To test whether EphrinA1 and EphA4 are involved in
I/R injury, the expression of EphrinA1 and EphA4 was examined in
stroke models in vitro and in vivo. OGD/R was used to
simulate ischemia-like conditions in vitro. After subjecting
the HBMECs to OGD/R for 12 or 24 h, western blotting demonstrated
that the expression of EphA4 and EphrinA1 was significantly
increased (Fig. 1A and B). Next, we
analyzed EphA4 and EphrinA1 expression alterations in the
ipsilateral cortical tissue following cerebral ischemia for 24, 48
and 72 h. Western blotting showed that the expression of EphA4 and
EphrinA1 was significantly increased in a time-dependent manner
(Fig. 1C and D), and the
immunohistochemistry staining also displayed higher levels of EphA4
and EphrinA1 expression following cerebral I/R for 72 h in the
ipsilateral ischemic brain regions (Fig.
1E).
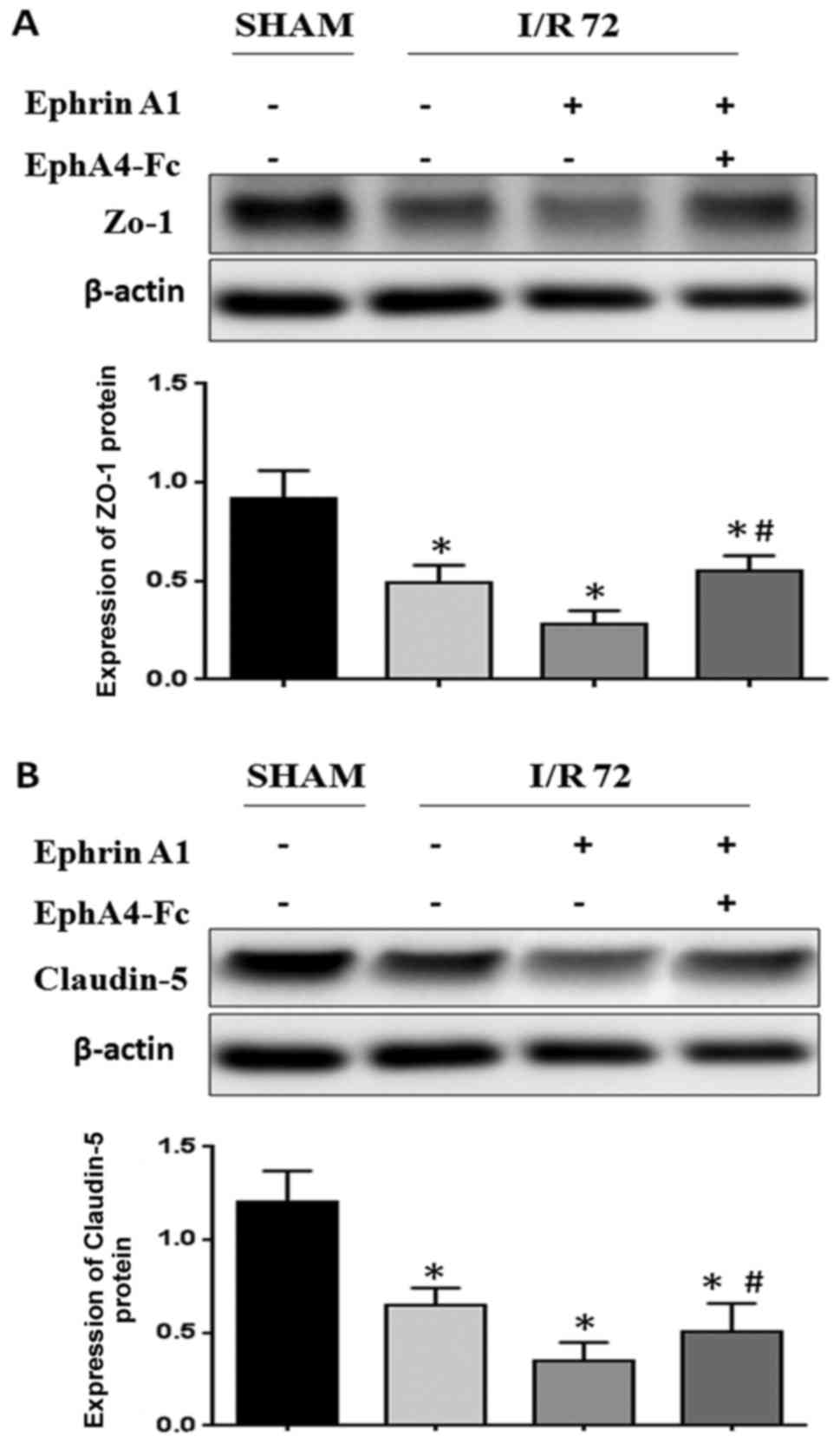
EphrinA1 contributes to BBB damage
through the EphA4 receptor
Since stroke is caused by the breakdown of BBB
integrity, we hypothesized that the upregulation of EphrinA1 may be
involved in the breakdown of BBB integrity following I/R injury
in vivo. We wanted to detect whether the weak BBB stability
following I/R was caused by EphrinA1 overexpression inducing
abnormal TJ protein regulation. We observed that the expression of
ZO-1 and Claudin-5 was decreased in EphrinA1-treated I/R mice
compared with the sham and I/R groups, thus indicating that the
decreased BBB stability in I/R mice could perhaps be due to
modulation of ZO-1 and Claudin-5 by EphrinA1. To determine whether
the upregulation of the EphA4 receptor was responsible for the
downregulation of ZO-1 induced by EphrinA1, we also used an EphA4
receptor inhibitor, EphA4-Fc, to examine the role of the
EphrinA1/EphA4 interaction in I/R mice. Surprisingly, our data
showed that the downregulation of ZO-1 and Claudin-5 in I/R mice
treated with EphrinA1 was reversed by the administration of
EphA4-Fc, which indicated that EphrinA1 contributed to the BBB
damage through the EphA4 receptor (Fig.
2A and B).
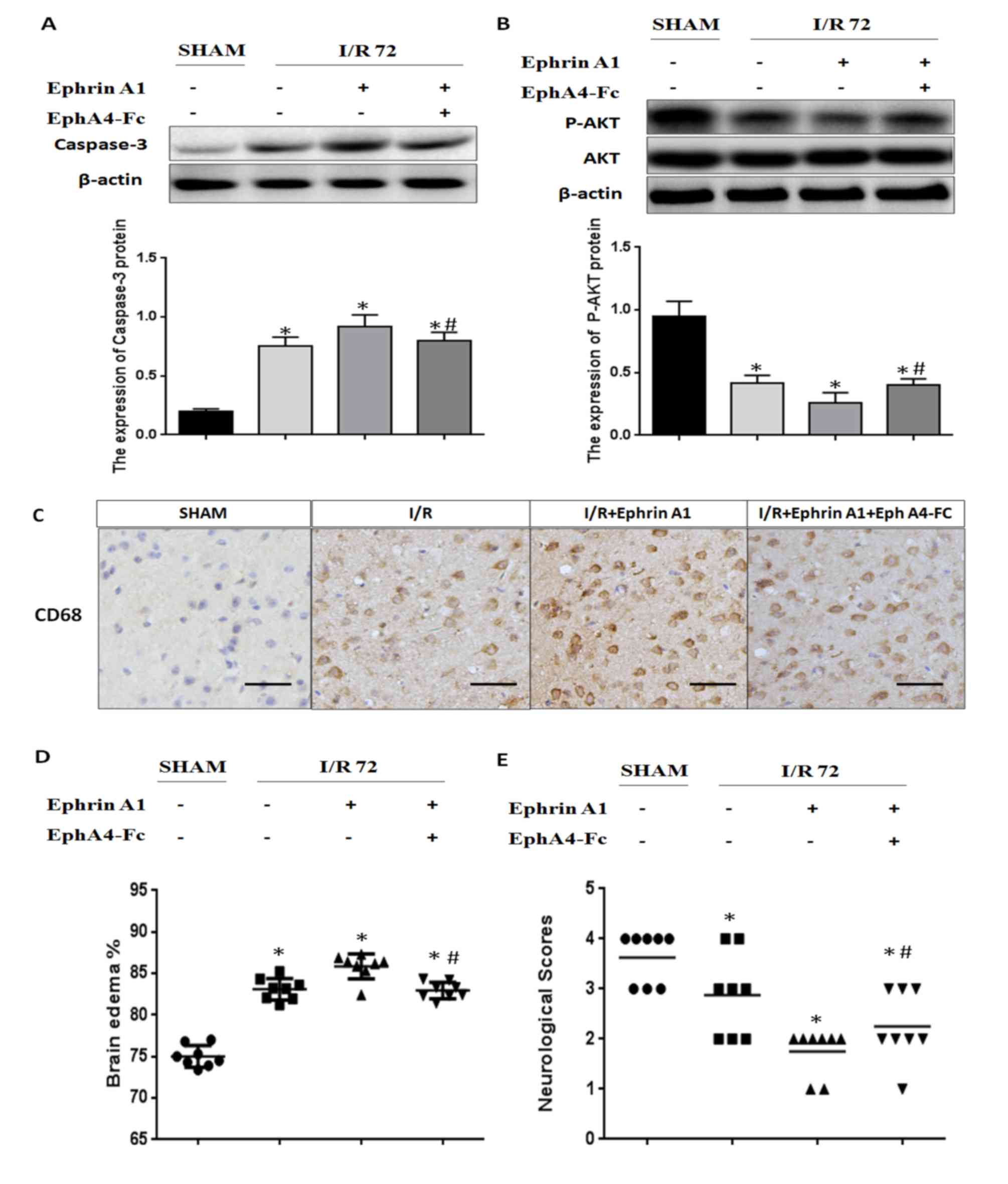
Cerebral I/R injury-induced
pro-apoptotic cell death signaling and brain damage were enhanced
by the EphrinA1/EphA4 signaling pathway
In order to investigate the molecular mechanisms
underlying the inactivation of the EphrinA1/EphA4 signaling pathway
in the I/R brain, we detected apoptotic signaling using immunoblot
analysis of caspase-3 and P-AKT. We found that the caspase-3
expression level was significantly increased in I/R mice treated
with EphrinA1. We also observed that I/R mice treated with EphrinA1
had significantly lower levels of P-AKT. Interestingly,
pretreatment with EphA4-Fc dramatically reduced apoptotic signaling
and promoted survival signaling in I/R mice stimulated with
EphrinA1, suggesting that ischemia-induced cell death is likely due
to the activation of the EphA4 receptor by EphrinA1 (Fig. 3A and B). Next, we assessed
inflammation levels, brain edema and neurological scores to
investigate the effect of the EphrinA1/EphA4 signaling pathway in
brain damage. Our data showed that EphrinA1 enhanced the
infiltration of CD68+ cells into the ipsilateral
hemisphere in 72-h I/R mice (Fig.
3C). In addition, in comparison with the sham and I/R groups,
EphrinA1-treated I/R mice exhibited significantly higher edema in
each brain hemisphere, accompanied by lower neurological scores
(Fig. 3D and E). Interestingly,
these effects of EphrinA1 were significantly attenuated by
EphA4-Fc, suggesting that the EphrinA1/EphA4 signaling pathway
contributes to brain damage in cerebral I/R injury.
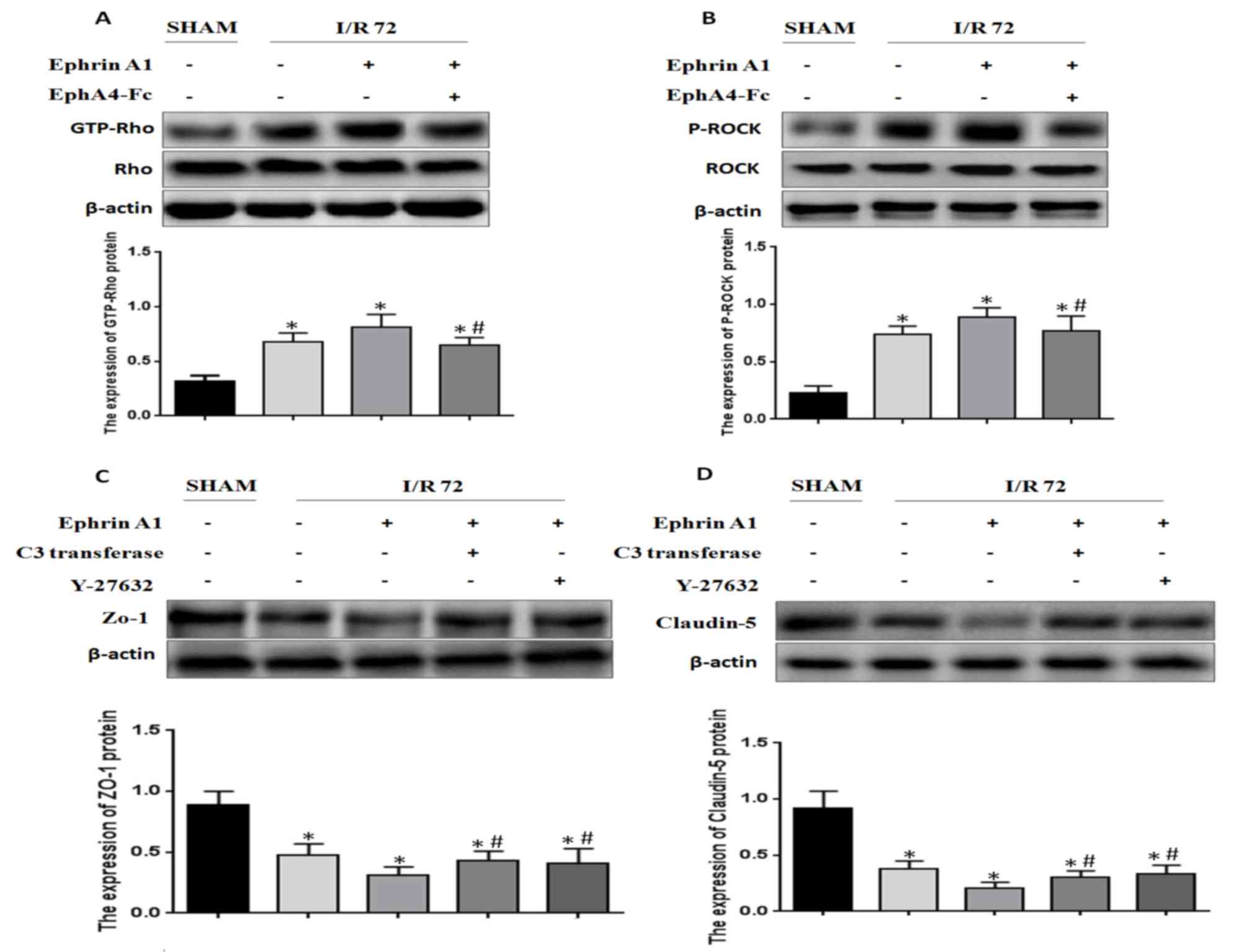
EphA4 activation by EphrinA1 induces
BBB damage through the Rho/ROCK pathway
It has been reported that Rho/ROCK signaling is
positively relevant to BBB damage induced by I/R. Based on this
finding, we wanted to detect whether EphrinA1 modulated RhoA
activity in I/R mice. We found that RhoA activity was enhanced
under the conditions of I/R, and EphrinA1 promoted RhoA activity in
I/R mice (Fig. 4A). In addition, the
phosphorylation of ROCK was also strengthened by EphrinA1 (Fig. 4B). Similarly, the effects of EphrinA1
were reversed by the administration of EphA4-Fc. Furthermore, we
explored whether EphrinA1 regulated the expression of ZO-1 and
Claudin-5 via activation of the Rho/ROCK signaling pathway. The
mice received pretreatment with C3 transferase or Y-27632 followed
by the administration of EphrinA1. The expression of ZO-1 and
Claudin-5 in each group was detected by western blotting. The
results demonstrated that C3 transferase or Y-27632 inhibited the
EphrinA1-induced downregulation of ZO-1 and Claudin-5 (Fig. 4C and D). Therefore, we conclude that
EphA4 activation by EphrinA1 induces BBB damage through the
Rho/ROCK pathway.
Discussion
Stroke is a major refractory disease with a high
rate of morbidity, disability and mortality, and it significantly
threatens human health and life expectancy worldwide. According to
previously published statistics, stroke markedly increases the risk
of dementia by 4 to 12 times (28).
Additionally, post-stroke dementia increases the risk of recurrent
stroke and mortality (29). At
present, there is a particular need to provide an. effective
therapeutic target for VD following ischemic stroke. Notably, the
breakdown of the brain vascular endothelium and disorders of tight
junctions induced by I/R injury lead to altered BBB permeability,
which promotes brain inflammation and edema, and even cognitive
impairment (30). However, the
signaling mechanisms associated with I/R-induced BBB damage remain
poorly understood. Previous studies have demonstrated that brain
microvascular endothelial cells are an important component of the
BBB; meanwhile, disorders of brain vascular permeability following
ischemic stroke contribute to secondary brain tissue damage
(31). In our research, we
highlighted the potential molecular mechanisms underlying BBB
damage in I/R. The primary finding of this study is that the
activation of the EphrinA1/EphA4 signaling pathway may enhance
I/R-induced-BBB damage. Inactivation of EphrinA1/EphA4 may be a
potential treatment for ischemic stroke and post-stroke
dementia.
At present, the role of the Eph/Ephrin system in the
context of the pathophysiology of stroke-induced BBB breakdown and
injury is unclear. It has been reported that Eph/Ephrin
interactions play a key role in embryonic development (32). However, increasing studies have
demonstrated that these receptors are able to promote some disease
processes. Moreover, it has been proven that the activation of the
EphA2 receptor by Eph ligands contributes to BBB damage and
neuronal death in ischemic stroke, suggesting a crucial role of
Eph/Ephrin signaling in these processes (26). Furthermore, inflammatory responses
participated in the complex ischemic cascade, contributing to the
damage to the BBB in ischemic stroke (24). Interestingly, it has been
demonstrated that EphrinA1/EphA4 signaling contributes to the
adhesion of monocytes to endothelial cells in vitro, which
may play a key role in inflammatory stimuli (23). Although EphrinA1 and EphA4 are known
to be expressed in brain microvascular endothelial cells, little is
known about whether the interaction between EphrinA1 and EphA4
plays an important role in the context of stroke-induced BBB damage
and injury. Therefore, the present study aimed to evaluate the
interaction between the EphA4 receptor and EphrinA1 ligand in the
molecular mechanisms underlying ischemic stroke.
The data presented in this study revealed that the
expression of EphrinA1 and EphA4 was upregulated, secondarily to
the stimulation of GD for 12 and 24 h. Next, these results were
validated in vivo, and a similar trend in EphrinA1 and EphA4
expression was also observed after MCAO for 24, 48 and 72 h, and
the immunohistochemistry staining in the ipsilateral ischemic brain
regions confirmed these results. Furthermore, we investigated the
role of the interaction between EphrinA1 and EphA4 in BBB damage
after stroke by evaluating the levels of the tight junction
proteins ZO-1 and Claudin-5. We found that EphrinA1-treated I/R
mice exhibited significantly lower levels of ZO-1 and Claudin-5
protein compared with the sham and I/R groups. However, EphA4
silencing by EphA4-Fc abolished the EphrinA1-mediated
downregulation of TJ proteins. These results suggested that the
upregulation of EphrinA1 contributed to BBB damage through the
EphA4 receptor.
Considering that cellular apoptosis plays a key role
in brain damage after ischemic stroke, we explored the effect of
the EphrinA1/EphA4 signaling pathway on the levels of apoptotic
proteins. Apoptosis-related proteins (Caspase-3 and p-Akt) were
measured by western blotting to assess apoptotic signaling.
Increased caspase-3 and decreased P-AKT protein expression was
observed in EprinA1-treated I/R groups compared with the sham and
I/R groups, which was attenuated dramatically by pretreatment with
EphA4-Fc. In addition, we hypothesized that inhibition of the
EphrinA1/EphA4 signaling pathway exhibited the potential to improve
the outcome of ischemic stroke in mice. Surprisingly, we found that
the EphrinA1-treated I/R mice had significantly enhanced
post-stroke edema and depraved neurological outcomes when compared
to sham and I/R mice, but EphA4-Fc was demonstrated to block
augmented brain injury via the administration of EphrinA1 in I/R
mice. Similarly, we observed that activation of the EphrinA1/EphA4
signaling pathway significantly facilitated inflammatory
infiltrates, which was consistent with a possible pathological
mechanism in I/R mice.
It is widely known that the Rho-kinase pathway plays
important roles in many cellular functions, including contraction,
motility, proliferation, and apoptosis (33). Increasing studies have reported that
the excessive activity of Rho-kinase exhibits the capacity to
induce oxidative stress and inflammatory response, which promotes
the development of cerebrovascular diseases (34). And a previous study demonstrated that
inhibition of Rho-kinase after ischemic stroke improved BBB
stability (35). Next, we wanted to
address whether the Ephrin A1/EphA4 interaction induced the
activation of Rho and ROCK. Interestingly, we observed enhanced
expression of GTP-RhoA and P-ROCK in EphrinA1-treated I/R mice,
which was also attenuated by the EphA4-Fc. Finally, we analyzed
whether EphrinA1/EphA4 exacerbated BBB damage via the Rho/ROCK
pathway. As expected, C3-transferase and Y27632 significantly
blocked the EphrinA1-mediated enhancement of ZO-1 and Claudin-5
degradation.
Our study identified the pathological effects of the
EphrinA1/EphA4 signaling pathway in ischemic stroke pathology. The
overexpression of Ephrin-A1 and EphA4 contributes to the damage to
the BBB and brain injury through the Rho/ROCK signaling pathway. We
advocate that inhibition of the EphrinA1/EphA4 signaling pathway in
the early stages following ischemic stroke may alleviate brain
damage and neuronal loss. Hopefully, the remission of ischemic
stroke via the inhibition of BBB damage may restrain the generation
and development of VD.
Acknowledgements
Not applicable.
Funding
This study was supported by a general project grant
from the Nanjing Military Region (grant no. 14MS015).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FBC and ZYL designed the study, performed the
experiments, acquired the data and wrote the manuscript. WP, ZQG,
HOY and TJY helped perform the experiments in the stroke model.
SBD, ZKC and BZ performed the immunohistochemistry and western blot
analysis. LJM assisted with the cell culture experiments and
critically reviewed the final manuscript. ZYC designed the study,
wrote the manuscript and finally reviewed the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Medical Ethics
Committee for Animal Care and Use at The 102nd Hospital of The
People's Liberation Army.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BBB
|
blood-brain barrier
|
|
VD
|
vascular dementia
|
|
HBMEC
|
human brain microvascular endothelial
cells
|
|
TJ
|
tight junctions
|
|
MCAO
|
middle cerebral artery occlusion
|
References
|
1
|
Plassman BL, Langa KM, Fisher GG, Heeringa
SG, Weir DR, Ofstedal MB, Burke JR, Hurd MD, Potter GG, Rodgers WL,
et al: Prevalence of dementia in the United States: The aging,
demographics and memory study. Neuroepidemiology. 29:125–132. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Erkinjuntti T: Diagnosis and management of
vascular cognitive impairment and dementia. J Neural Transm Suppl.
1–109. 2002.
|
|
3
|
Iadecola C: The pathobiology of vascular
dementia. Neuron. 80:844–866. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bowler JV: Vascular cognitive impairment.
Stroke. 35:386–388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nyenhuis DL and Gorelick PB: Vascular
dementia: A contemporary review of epidemiology, diagnosis,
prevention and treatment. J Ame Geriat Soc. 46:1437–1448. 1998.
View Article : Google Scholar
|
|
6
|
Farooq MU and Gorelick PB: Vascular
cognitive impairment. Curr Atheroscler Rep. 15:3302013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jacquin A, Binquet C, Rouaud O,
Graule-Petot A, Daubail B, Osseby GV, Bonithon-Kopp C, Giroud M and
Béjot Y: Post-stroke cognitive impairment: High prevalence and
determining factors in a cohort of mild stroke. J Alzheimers Dis.
40:1029–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Imfeld P, Bodmer M, Schuerch M, Jick SS
and Meier CR: Risk of incident stroke in patients with Alzheimer
disease or vascular dementia. Neurology. 81:910–919. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pendlebury ST and Rothwell PM: Prevalence,
incidence and factors associated with pre-stroke and post-stroke
dementia: A systematic review and meta-analysis. Lancet Neurol.
8:1006–1018. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang Y and Rosenberg GA: Blood-brain
barrier breakdown in acute and chronic cerebrovascular disease.
Stroke. 42:3323–3328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kamada H, Yu F, Nito C and Chan PH:
Influence of hyperglycemia on oxidative stress and matrix
metalloproteinase-9 activation after focal cerebral
ischemia/reperfusion in rats: Relation to blood-brain barrier
dysfunction. Stroke. 38:1044–1049. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khatri R, McKinney AM, Swenson B and
Janardhan V: Blood-brain barrier, reperfusion injury and
hemorrhagic transformation in acute ischemic stroke. Neurology. 79
13 Suppl 1:S52–S57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weekman EM and Wilcock DM: Matrix
metalloproteinase in blood-brain barrier breakdown in dementia. J
Alzheimers Dis. 49:893–903. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rochfort KD and Cummins PM: The
blood-brain barrier endothelium: A target for pro-inflammatory
cytokines. Biochem Soc Trans. 43:702–706. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Blanchette M and Daneman R: Formation and
maintenance of the BBB. Mech Dev. 138:8–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Haseloff RF, Dithmer S, Winkler L, Wolburg
H and Blasig IE: Transmembrane proteins of the tight junctions at
the blood-brain barrier: Structural and functional aspects. Semin
Cell Dev Biol. 38:16–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Abbott NJ, Patabendige AA, Dolman DE,
Yusof SR and Begley DJ: Structure and function of the blood-brain
barrier. Neurobiol Dis. 37:13–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stamatovic SM, Keep RF and Andjelkovic AV:
Brain endothelial cell-cell junctions: How to ‘open’ the blood
brain barrier. Curr Neuropharmacol. 6:179–192. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sandoval KE and Witt KA: Blood-brain
barrier tight junction permeability and ischemic stroke. Neurobiol
Dis. 32:200–219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lucke-Wold BP, Logsdon AF, Turner RC,
Rosen CL and Huber JD: Aging, the metabolic syndrome and ischemic
stroke: redefining the approach for studying the blood-brain
barrier in a complex neurological disease. Adv Pharmacol.
71:411–449. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lisabeth EM, Falivelli G and Pasquale EB:
Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol.
5:a0091592013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ieguchi K: Eph as a target in
inflammation. Endocr Metab Immune Disord Drug Targets. 15:119–128.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jellinghaus S, Poitz DM, Ende G, Augstein
A, Weinert S, Stütz B, Braun-Dullaeus RC, Pasquale EB and Strasser
RH: Ephrin-A1/EphA4-mediated adhesion of monocytes to endothelial
cells. Biochim Biophys Acta. 1833:2201–2211. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gliem M, Schwaninger M and Jander S:
Protective features of peripheral monocytes/macrophages in stroke.
Biochim Biophys Acta. 1862:329–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moskowitz MA, Lo EH and Iadecola C: The
science of stroke: Mechanisms in search of treatments. Neuron.
67:181–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Thundyil J, Manzanero S, Pavlovski D,
Cully TR, Lok KZ, Widiapradja A, Chunduri P, Jo DG, Naruse C, Asano
M, et al: Evidence that the EphA2 receptor exacerbates ischemic
brain injury. PLoS One. 8:e535282013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Choi JS, Kim SJ, Shin JA, Lee KE and Park
EM: Effects of estrogen on temporal expressions of IL-1beta and
IL-1ra in rat organotypic hippocampal slices exposed to
oxygen-glucose deprivation. Neurosci Lett. 438:233–237. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Khedr EM, Hamed SA, El-Shereef HK, Shawky
OA, Mohamed KA, Awad EM, Ahmed MA, Shehata GA and Eltahtawy MA:
Cognitive impairment after cerebrovascular stroke: Relationship to
vascular risk factors. Neuropsychiatr Dis Treat. 5:103–116.
2009.PubMed/NCBI
|
|
29
|
del Ser T, Barba R, Morin MM, Domingo J,
Cemillan C, Pondal M and Vivancos J: Evolution of cognitive
impairment after stroke and risk factors for delayed progression.
Stroke. 36:2670–2675. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van de Haar HJ, Burgmans S, Hofman PA,
Verhey FR, Jansen JF and Backes WH: Blood-brain barrier impairment
in dementia: Current and future in vivo assessments. Neurosci
Biobehav Rev. 49:71–81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Borlongan CV, Rodrigues AA Jr and Oliveira
MC: Breaking the barrier in stroke: What should we know? A
mini-review. Curr Pharm Des. 18:3615–3623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hafner C, Schmitz G, Meyer S, Bataille F,
Hau P, Langmann T, Dietmaier W, Landthaler M and Vogt T:
Differential gene expression of Eph receptors and ephrins in benign
human tissues and cancers. Clin Chem. 50:490–499. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Julian L and Olson MF: Rho-associated
coiled-coil containing kinases (ROCK): Structure, regulation and
functions. Small GTPases. 5:e298462014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shin HK, Salomone S and Ayata C: Targeting
cerebrovascular Rho-kinase in stroke. Exp Opin Ther Targets.
12:1547–1564. 2008. View Article : Google Scholar
|
|
35
|
Gibson CL, Srivastava K, Sprigg N, Bath PM
and Bayraktutan U: Inhibition of Rho-kinase protects cerebral
barrier from ischaemia-evoked injury through modulations of
endothelial cell oxidative stress and tight junctions. J Neurochem.
129:816–826. 2014. View Article : Google Scholar : PubMed/NCBI
|