Introduction
Velo-cardio-facial syndrome [VCFS; Online Mendelian
Inheritance in Man (OMIM) cat. no. 192430] is a multiple
malformation syndrome, which is characterized by highly variable
clinical features, including cleft palate, cardiac anomalies,
atypical facial development and cognitive and neuropsychological
difficulties (1,2). The first case of VCFS was described in
1955 by Eva Sedlačková. DiGeorge described the association between
VCFS and thymic aplasia, hypoparathyroidism and congenital heart
disease in children in 1968. While in 1978 R. J. Shprintzen
presented 12 cases of VCFS, including a family of one, and
established it as a distinct inherited genetic disorder (3). In 90% of patients with VCFS, a de
novo variably sized deletion at chromosome 22q11.2 is
responsible for the syndrome (4).
VCFS occurs in between 1 in every 4000 and 7000
births (3). The condition has been
previously described by several physicians and has been given
several different names including, VCFS, Shprintzen syndrome,
DiGeorge syndrome (DGS), DiGeorge sequence, CATCH 22, deletion
22q11 syndrome, Cayler syndrome and conotruncal anomaly face
syndrome. VCFS is the fourth most common type of congenital anomaly
worldwide. However, in Romania there is no comprehensive data on
the prevalence of the disease.
The present study reports the case of a newborn male
with keilopalatoschisis, dysmorphic face, heart anomalies, genital
hypoplasia and varus equinus, including the clinical data and
cytogenetic information. The cytogenetic evaluation revealed an
unusual, unbalanced translocation involving chromosomes 22 and 15
in a karyotype with 45 chromosomes, which is a rare
rearrangement.
Materials and methods
The present study complied with the Declaration of
Helsinki and was approved by the institutional ethics committee of
the Victor Babeș University of Medicine and Pharmacy (Timișoara,
Romania). Written informed consent was obtained from the legal
guardian of the child for the use of their case details and
associated images in the present study.
The present paper presents the case of a male child
with VCFS. Physical examination was conducted in order to identify
anatomical problems. Cardiac disorders have been identified,
according to clinical and paraclinical criteria, by thoracic
radiography, ECG and Ecocardiography. ECG and Echocardiography were
recorded with a Schiller and ESAOTE machine, respectively. Oxygen
saturation was assessed with a pulse oximeter.
Cytogenetic analysis
Standard lymphocyte cytogenetic analysis was
performed using peripheral blood followed by GTG-banding at the
550-band level (5). A number of 20
metaphases were analyzed by two independent observers using a Nikon
ECLIPE 55i trinocular microscope. For karyotyping a dedicated Lucia
Karyo software was used.
Fluorescence in situ hybridization
(FISH) analysis
FISH was performed using the Metasystem XL Probes
for Microdeletions 22q11.2 TUPLE1 DiGeorge sample (HIRA-HIR histone
cell cycle regulation defective homolog A)-red (120 kb), and SHANK3
control sample 22q13-green (40 kb) (cat. no. D-6404-050-RG). The
FISH analyses were performed by two independent observers using a
Nikon Eclipse 600 microscope equipped with a standard fluorescence
isothiocyanate filter. The photographs were captured using Kodak
Ektachrome 400 film.
The results of the cytogenetic and FISH analyses are
described further according to the International System for Human
Cytogenomic Nomenclature 2016 (6).
The control sample was obtained from a male, 7 months old patient,
admitted to the Onco-Hematology department of the Louis Turcanu
hospital (Timisoara, Romania; August 2013).
Results
Case presentation
The patient was born by caesarean section at 37
weeks and 5 days of gestation, after an uncomplicated pregnancy to
a healthy 18-year-old woman. It was the first pregnancy for the
nonconsanguineous healthy couple. At birth the child was 3,200 g
[-0.57 standard deviation (SD)], 49 cm in length (−0.9 SD), had a
head circumference of 30 cm (−4.72 SD) and the Apgar score was 6.
After birth the patient was artificially fed and his weight gain
was impaired. The mother denies taking any treatment during the
pregnancy and there are no reports of consanguinity or genetic
anomalies in the family history. The parents were examined in
detail and were not found to have any features of the syndrome, or
any history of reproductive health problems. The parents have
subsequently divorced and the mother has married an
African-American male and had another child (Fig. 1). An amniocentesis performed during
the second pregnancy revealed that the child had a normal
karyotype.
At the age of 3 h, the patient described in the
manuscript, was referred to a pediatric ward for evaluation of the
plurimalformative syndrome and poor neonatal adaptation. The
patient was hypertonic and had peripheral cyanosis. The clinical
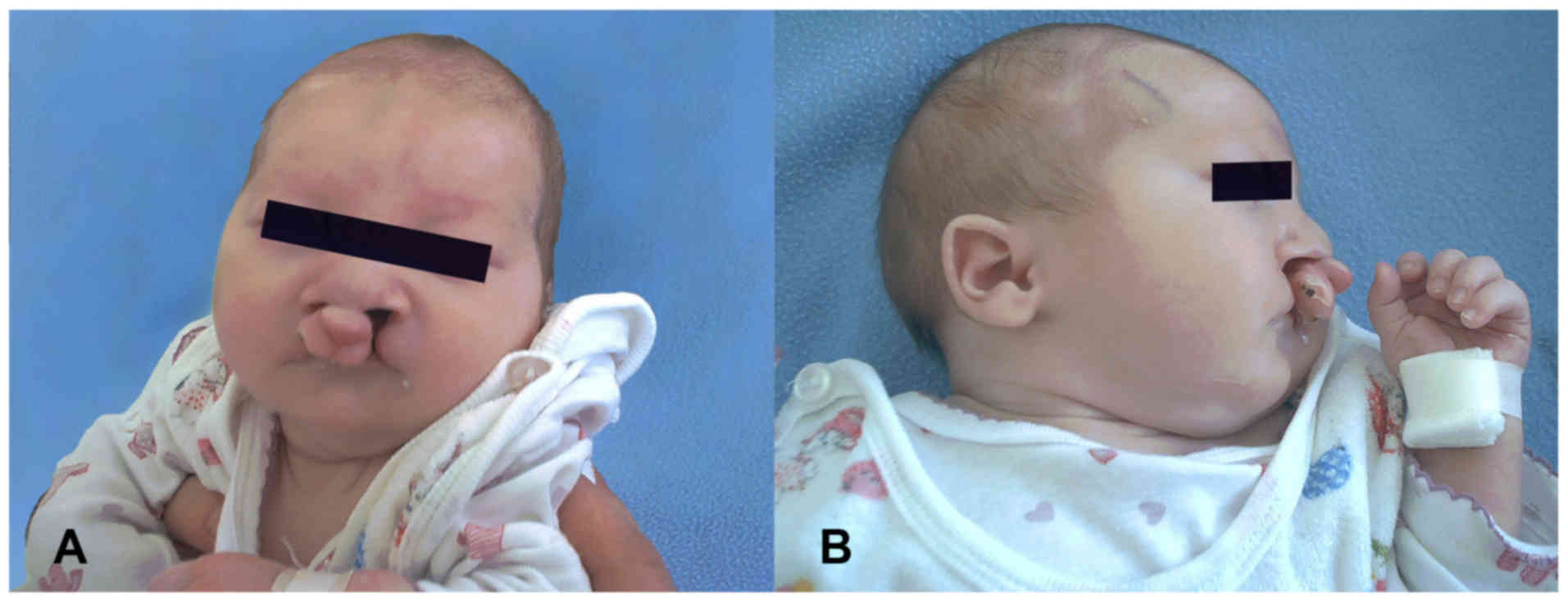
evaluation revealed microcephaly, a long and hypotonic face with
mild orbital hypertelorism, almond-shaped eyes, dark red rings
under the eyes, a prominent nasal bridge, a long but wide nose with
a bulbous nasal tip, flat cheekbones, down-turned corners of the
mouth, an overt cleft palate with velopharyngeal insufficiency,
micrognathia, small and low-set ears (Fig. 2A and B), cryptorchidism and clubfoot.
Cardiac examination revealed a grade II systolic murmur in the
upper left sternal border, as well as a grade II systolic murmur in
the lower left sternal border, which irradiated all over the
precordium. Pulmonary rales were revealed by auscultation.
Further biological investigation identified multiple
systemic and peripheral infections due to the patient's condition,
including anemia and hypogammaglobulinemia. Oxygen saturation was
impaired (78%) and an electrocardiogram revealed sinus rhythm,
right axis deviation and right atrial and ventricular hypertrophy.
Cardiopulmonary X-ray revealed a ‘boot shaped’ heart, a
cardiothoracic index of 0.67, increased prominence of the pulmonary
artery and decreased vascular markings (Fig. 3) An echocardiograph revealed a
ventricular septal defect, hypertrophy of the right ventricle,
overriding of the aorta and pulmonary artery stenosis, which
confirmed the diagnose of Tetralogy of Fallot (Fig. 3A-C).
The hypertonia, clonus and incomplete archaic
reflexes revealed a perinatal hypoxic-ischemic injury. A
transfontanellar ultrasound was performed when the patient was 2
days old; it identified perinatal hypoxic-ischemic injury with
intraventricular hemorrhage. The patient's audiometry was normal.
These results led to a diagnosis of VCFS.
The patient had multiple subsequent hospital
admissions due to recurrent pulmonary infection, secondary to
aspiration syndrome and their Tet spells were reported as severe.
Cardiac and oral surgery were not performed as consent was not
obtained from the parents. The patient was followed up until the
age of 1.5 years when they succumbed to the disease.
Cytogenetic analysis
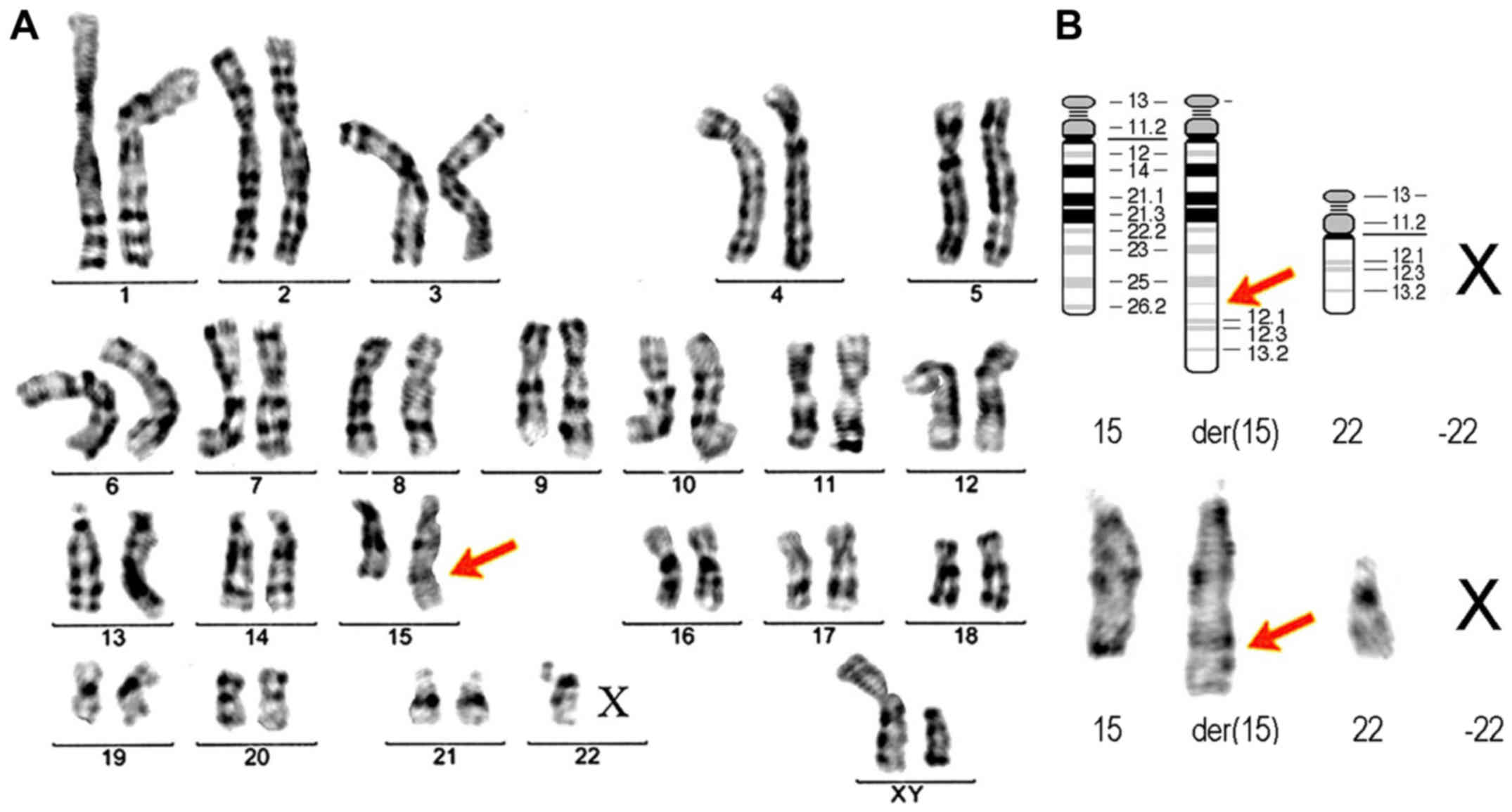
Revealed a translocation involving chromosomes 15
and 22 in a 45 chromosome karyotype; additional material was
observed in the long arm of chromosome 15, and one chromosome 22
was missing (Fig. 4). The karyotype
of the patient was given as
45,XY,-22,der(15),t(15;22)(q26.2;q11.2)dn. The derivate der(15)
replaced a normal chromosome 15 and the homologous chromosome 22
was lost. The karyotype confirmed the etiology of the case; 22
monosomy with unbalanced translocation of the genetic material from
the 22 chromosome (22q12-ter band), to the 15 chromosome. The
translocation took place with a 22q11.2 deletion. The parental
karyotypes were observed to be normal.
Fluorescence in situ hybridization
(FISH) analysis
Due to the clinical findings, FISH was performed on
metaphase chromosomes using a probe specific for the DGS critical
region (TUPLE). A single red signal from the 22q11.2 probe was
observed on the normal chromosome 22 (Fig. 5A). Whereas, 2 green signals from the
22q13.33 probe were observed on the subtelomere of the normal
chromosome 22 and the translocated chromosome. As the 22q11.2 red
probe has a size of 120 kb (4440 kDa), it is known that the
deletion has a minimum of 120 kb. The results of the FISH analysis
indicated
45,XY,-22,der(15),t(15;22).ishdel(22)(q11.2q11.2)(D22S451-) for the
patient and 46,XY.ish q22.11.2(D22S451×2) for the control
probe.
Differential diagnosis
A differential diagnosis was performed to consider
several diseases, as the phenotypic manifestations of VCFS are
pleiotropic. Kabuki syndrome (OMIM cat. no. 147920) was considered
due to the observation of cleft palate, cardiac anomaly and
hypotonia, however it was excluded due to the facial appearance and
small ears. Other conditions were excluded due to an incorrect
phenotype, including fetal alcohol syndrome (due to the heart
anomaly and cleft palate), Smith-Lemli-Opitz syndrome (OMIM cat.
no. 270400; due to the cleft palate), Alagille syndrome (OMIM cat.
no. 118450; due to the congenital heart disease), VATER syndrome
(OMIM cat. no. 192350) and Goldehar syndrome (OMIM cat. no.
%164210). Recent medical advancements regarding VCFS suggest that
patients previously diagnosed with Pierre Robin Sequences (PRS;
OMIM cat. no. %261800) and CHARGE association (OMIM cat. no.
214800) should undergo further clinical and cytogenetic evaluation.
The present case did not have CHARGE association (no coloboma in
the eyes, no choanal atresia and no deafness), or PRS (no
glossoptosis). VCFS, as multisystemic syndrome, is difficult to
identify as a minimum of 30 different symptoms have been associated
with the 22q11 deletion. Case by case evaluation is even more
difficult, as the majority of symptoms are not present in all
individuals who have VCFS (https://www.genome.gov/25521139/learning-about-velocardiofacial-syndrome/).
Discussion
VCFS is caused by a microdeletion at chromosome
22q11.2 and is the most common type of contiguous gene syndrome in
humans (4). Many healthcare
professionals now refer to patients with VCFS as having a 22q11.2
deletion. The deleted region of the chromosome contains information
for the development of organs from the third and fourth pharyngeal
pouches, during the 12th week of gestation (7).
No correlations have been found between the position
of the deleted fragment and the genes located at 22q11.2, which are
included in Table I (6). The first large study on VCFS evaluated
156 cases with deletions localized at 22q11, and no correlations
were observed between the size of the deletion and the phenotype
(4). In the present case, the
deletion was a minimum of 120 kb in size, as this is the size of
the TUPLE1 22q11.2 orange probe used for the FISH analysis. VCFS is
a complex disorder with a variable phenotype and penetrance; it is
thought that several genes in the commonly deleted region
contribute to the phenotype. VCFS transmission has a pattern of
autosomal dominant inheritance (8).
When one parent has VCFS, the probability of their children having
the syndrome is about 50% for each birth. However, previous
research has shown that VCFS is only inherited in 10 to 15% of
cases. In the present case the parents were clinically healthy,
with normal karyotypes and no signs of VCFS. It is most probable
that a de novo translocation occurred, with a consecutive
22q11.2 deletion (9).
 | Table I.Unbalanced translocations involving
deletion 22q11.2. |
Table I.
Unbalanced translocations involving
deletion 22q11.2.
| Translocation | De
novo/hereditary | Author, study | Abnormality | (Refs.) |
|---|
|
45,XY,-22,der(15),t(15;22)(q26.2;q11.2) | De novo | Present case | VCFS |
|
| 45,XX,-3,-22,
+der(3),t(3;22)(p25;q11) | De
novo/IVF | Faed et al,
1987 | DGS | (15) |
|
46,XY,-15,+der(22),t(15;22)(q13;q11) | Paternal | Van Hove et
al, 1992 | DGS + duplication
of 22q11 | (12) |
|
46,XY,t(15;22)(q22;q13) | De novo | Fryns, 1993 | DGS | (13) |
|
45,XX,der(4)t(4;22)(p16.3;q11.2),-22 | Maternal | Reddy et al,
1996 | DGS +
Wolf-Hirschhorn deletions | (16) |
|
46,XX,der(15),t(15;22)(p11.2;q11.2),-22 | De novo | Jaquez et
al, 1997 | DGS + VCGS | (11) |
|
t(9;22)(q34.3;q11.2) | Paternal | McGoey et
al, 2009 | DGS + 9q
subtelomeric deletion | (19) |
|
45,XY,der(3)t(3;22)(p25;q11),-22 | De novo | Dundar et
al, 2010 | VCFS + 3p
deletion | (17) |
|
45,XX,der(6)t(6;22)(p25.3;q11.21),-22 | De
novo/FIV | Gollo Dantas et
al, 2016 | DGS | (10) |
| 46,XX,r(22); | De novo | Kashevarova, et
al, 2018 | 22q13.32-q13.33
deletion | (20) |
There are many different translocations between
chromosome 22q11.2 and certain other chromosomes. This is due to
the presence of a region that contains 8 chromosome-specific
low-copy repeats within 22q11.2, which is a highly conserved DNA
sequence (>96%), which mediates non-allelic homologous
recombination, resulting in chromosome 22 rearrangements (10).
The etiology of the present case (unbalanced
translocation from chromosome 22 to chromosome 15 with 22 monosomy)
is very rare. However, a previous study described one case with the
karyotype 46,XX,der(15),t(15;22)(p11.2;q11.2),-22 and a clinical
appearance suggestive of DGS/VCFS, without a cleft palate (11). Chromosome 22 monosomy was observed,
as in the present case, but with a different breaking point; the
terminal fragment of chromosome 22 was translocated onto the short
arm of chromosome 15.
In another case with the karyotype
46,XY,-15,+der(22), t(15;22)(q13;q11), the patient presented
typical manifestations of a deletion of 15pter-q13 (severe
hypotonia and lethargy) and also typical signs of a 22q11-ter
duplication (hypertelorism, down-slanting small palpebral fissures,
preauricular tags and long philtrum) (12). That case had chromosome 15 monosomy,
and the unbalanced translocation was inherited from the father who
had a reciprocal translocation with a different point of rupture on
chromosome 15. One case of a reciprocal translocation
t(15;22)(q22;q13) without either monosomy 15 or 22 with
fronto-nasal malformation was also previously reported (13). To the best of our knowledge, only 7
live-born infants with mosaicism for monosomy of chromosome 22
associated with a unique facial appearance, similar to those with
DGS, have been previously described (14).
There have been some special cases of DGS/VCFS
occurring de novo in a patient conceived via in vitro
fertilization (IVF), in which translocation t(3;22)(p25;q11)
(15) and translocation
t(6;22)(p25.3;q11.21) (10) have
been identified. In patients with these translocations, the loss of
the proximal 22q region usually results in 22q11.2 deletion
syndrome, associated with monosomy of chromosome 22. The
rearrangements could be due to the manipulation of the embryo, or a
sporadic event unrelated to IVF (10).
In certain cases, translocations involving
chromosome 22 and another autosom can be phenotypically associated
with a combination of specific signs for DGS/VCFS and another
anomaly. These other anomalies may include translocation
t(4;22)(p16.3;q11.2) with Wolf-Hirschhorn deletions (16), translocation t(3;22)(p25;q11) with 3p
deletion (17), translocation
t(18;22)(p11.2;q11.2) with 18p deletion (18), translocation t(9;22)(q34.3;q11.2)
with 9q subtelomere deletion (19)
or translocation t(15;22)(q13;q11) with 22q11 duplications
(12) (Table I).
There have also been previously reported cases of
22q11 deletion syndrome, due to unequal segregation of balanced
parental translocations between chromosome 22 and another autosomal
chromosome. These may either be a paternal balanced reciprocal
translocation t(9;22)(q34.3;q11.2) (19) or maternal balanced reciprocal
translocations t(4;22)(p16.3;q11.2),-22 (16) or t(18;22)(pl1.2;q11.2) (18) (Table
I).
Recently (20), a
case was reported with the deletion del 22q13.32-q13.33, which was
associated with a ring chromosome r(22); its instability led to a
monosomy for chromosome 22 in mosaic as detected by FISH.
Less than 1% of all 22q11 deletions are the result
of an unbalanced translocation, in which chromosome 22 and another
chromosome are involved. In the present case the translocation
involved chromosomes 15 and 22. The chromosome 15 derivate had
genetic material from the chromosome 22q11 band as far as the
telomeres on its terminal end, in the 15q26 band. The 22-breaking
point was in band 22q11, the critical region for DGS 1, and the
pericentromeric region of chromosome 22 has been lost. This
cytogenetic aspect correlates with the phenotypic aspect, which is
suggestive of VCFS. The family underwent genetic counseling,
including nature, type of transmission and clinical and social
aspects of this anomaly. About 93% of all patients have a de
novo deletion of 22q11, while 7% have inherited the 22q11
deletion from a parent (8). The risk
of recurrence in a patient's siblings is relatively low as it was a
de novo translocation. Although the precise risk of germinal
mutations cannot be determined; these results have implications for
genetic counseling because there is a risk of transmission by germ
cells carrying the deletion, even when parents present a normal
karyotype in their blood cells (21).
Most patients with VCFS have a large (>3 Mb)
genomic deletion in chromosome 22q11, which includes the DiGeorge
critical region; this region is deleted in 90% of DGS patients with
a detectable deletion (4). In
familial cases the smaller deletions were found to be predominant
(22). A significant number of these
patients (~10%) have no demonstrable chromosomal deletion (23). Some families have previously
presented with classic features of DGS without evidence of a
chromosomal deletion at 22q11, but with specific TBX1 mutations,
including 2 missense mutations and a frameshift mutation (15,16,24,25). The
Tbox transcription factor (TBX1) gene, located at 22q11.21
is considered the major candidate for 22q11.2 deletion syndrome
(26), as it is associated with
cardiovascular defects and craniofacial and dental features, which
were also present in the patient in the current study. The T-box 1
protein acts as a transcription factor and appears to be necessary
for the normal development of muscles and bones in the face and
neck, large arteries that carry blood out of the heart, structures
in the ear and glands such as the thymus and parathyroid
(https://ghr.nlm.nih.gov/gene/TBX1).
At present 2 genes (COMT and TBX1) are associated
with VCFS. However, not all the genes that cause VCFS have been
identified. (https://www.genome.gov/25521139/learning-about-velocardiofacial-syndrome).
A multidisciplinary evaluation involving healthcare
professionals from specialties including, genetics, plastic
surgery, speech pathology, otorhinolaryngology, cardiology, cardiac
surgery, child development and psychology, neurology, orthopedics,
hematology, immunology, endocrinology and pediatrics is often
necessary for a successful clinical diagnosis of VCFS.
Non-characteristic features are common in deletion 22q11. Many
treatable conditions may be prematurely diagnosed and the
pathological features may accumulate over time (27). The severity and number of problems
varies from patient to patient, resulting a combination of
impairments and disabilities (28).
The absence of typical facial features in African-Americans
patients with the 22q11.2 deletion may result in a decreased
diagnosis of the syndrome within this population, and may delay the
implementation of palliative care, cognitive remediation and
recurrence risk counseling (8). This
information could be relevant in the future as in the present study
the mother's second husband was an African-American.
Aside from cleft palate, there are at least 184
other anomalies, including other abnormal facial characteristics,
commonly associated with VCFS. It is considered that VCFS is the
most frequent clefting syndrome, and it occurs in 8.1% of children
with cleft palate (29). A rare VCFS
case with cleft palate, cardiac malformation and progressive
pancytopenia has also been reported (30). The patient in the current study had a
complete palatal cleft, which is observed in 69% of patients with
VCFS. Due to feeding difficulties and severe dysphagia the patient
needed a nasogastric tube for enteric feeding; this is observed in
50% of all patients with VCFS. The patient also had tetralogy of
Fallot, which is considered the most common congenital heart
disease in VCFS (31). Cardiac
defects are found in 84% of patients with VCFS and are the main
cause of morbidity and mortality (32). The craniofacial findings were quite
variable.
Although most patients have a history of hypotonia
in infancy and learning disabilities (33), specific neurological manifestations
are rare. Seizures were seen in some patients and were most often
associated with hypocalcemia. The patient in the current study
presented with neurological signs including hypertonia, clonuses
and incomplete archaic reflexes.
The CATCH 22 acronym (C, cardiac anomalies; A,
abnormal faces; T, thymus hypoplasia; C, cleft palate; H,
hypocalcemia; 22, affected chromosome) was suggested as an
alternative name for the syndrome (34). Among pathophysiological disorders,
DGS is classified as an isolated T cell deficiency, due to impaired
development of the thymus gland, with recurrent bacterial, viral
and fungal infections (35). Both
hypocalcemia, which occurs due to partial or complete absence of
the parathyroid gland, and thymus hypoplasia were absent in the
present case.
Genetic counseling may be very difficult and
complex. Chromosome 22 at band q11.2 and chromosome 15 at band
q11q13 are considered unstable regions (36). The genetic risk of the family having
children with congenital anomalies exists on every future
pregnancy. It was recommended that the mother should receive
invasive prenatal diagnosis in all future pregnancies (37). The mother approached the authors for
an amniocentesis during her next pregnancy, which revealed a normal
karyotype, and a healthy child was born (Fig. 1).
In conclusion, a translocation involving chromosome
22 in a karyotype with 45 chromosomes is a rare event and, to the
best of our knowledge, this has not been previously reported
involving chromosomes 15q and 22q. The major malformations observed
in the present case suggested the diagnosis, which was confirmed by
the unbalanced t(15;22) translocation with 22q11.2 deletion
revealed by standard karyotyping and FISH. Genetic diagnosis is
essential to enable a successful diagnosis and genetic counseling
for the family.
Acknowledgements
The authors would like to thank their colleagues
from the Emergency Clinical Hospital for Children ‘Louis Turcanu’
(Timisoara, Romania) for their cooperation during the case
evaluation.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during the study are
included in this published article.
Authors' contributions
CG performed the cytogenetic analysis, the genetic
counseling and wrote the first draft of the manuscript. IM, DH and
MV revised and improved the first draft of the manuscript, made
substantial contributions by collecting the data from the
literature included in Table I and
revising the manuscript critically for important intellectual
content. CP and LG performed the FISH analysis. CP was also the
second evaluator for cytogenetic analysis. GD performed the
cardiology evaluation. RS was the pediatrician who treated the
child in the clinic throughout his admissions. The mother's
pregnancy was monitored by GF and CF. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
The present study complied with the Declaration of
Helsinki and has been approved by the institutional ethics
committee of the Victor Babes University of Medicine and Pharmacy
(Timisoara, Romania). Written informed consent was obtained from
the legal guardian of the patient and from the parent of the child
who provided the control probe for the use of their clinical data
and associated images in the present study.
Patient consent for publication
The legal guardian of the child gave written consent
to publish the medical information associated with the patient.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
OMIM
|
Online Mendelian Inheritance in
Man
|
|
VCFS
|
velo-cardio-facial syndrome
|
|
FISH
|
fluorescence in situ
hybidization
|
|
PRS
|
Pierre Robin Sequences
|
|
DGS
|
DiGeorge syndrome
|
|
CATCH 22 acronym
|
(C=cardiac anomalies, A=abnormal
faces, T=thymus hypoplasia, C=cleft palate, H=hypocalcaemia,
2=affected chromosome)
|
|
IVF
|
in vitro fertilization
|
References
|
1
|
Shprintzen RJ, Goldberg RB, Lewin ML,
Sidoti EJ, Berkman MD, Argamaso RV and Young D: A new syndrome
involving cleft palate, cardiac anomalies, typical facies, and
learning disabilities: Velo-cardio-facial syndrome. Cleft Palate J.
15:56–62. 1978.PubMed/NCBI
|
|
2
|
Sandrin-Garcia P, Richieri-Costa A, Tajara
EH, Carvalho-Salles AB and Fett-Conte AC: Fluorescence in situ
hybridization (FISH) screening for the 22q11.2 deletion in patients
with clinical features of velocardiofacial syndrome but without
cardiac anomalies. Genet Mol Biol. 30:21–24. 2007. View Article : Google Scholar
|
|
3
|
Gothelf D, Frisch A, Michaelovsky E,
Weizman A and Shprintzen RJ: Velo-cardio-facial syndrome. J Ment
Health Res Intellect Disabil. 2:149–167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carlson C, Sirotkin H, Pandita R, Goldberg
R, McKie J, Wadey R, Patanjali SR, Weissman SM, Anyane-Yeboa K,
Warburton D, et al: Molecular definition of 22q11 deletions in 151
velo-cardio-facial syndrome patients. Am J Hum Genet. 61:620–629.
1997. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yunis JJ: High resolution of human
chromosomes. Science. 191:1268–1270. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McGowan-Jordan J, Simons A and Schmid M:
ISCN 2016. Publisher Karger; 2016, View Article : Google Scholar
|
|
7
|
McCance KL, Huether SE, Brashers VL and
Rote NS: Pathophysiology. The biologic basis for disease in adults
and children. 6th. Mosby Elsevier; Missouri: 2010
|
|
8
|
McDonald-McGinn DM, Tonnesen MK,
Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS and Zackai EH:
Phenotype of the 22q11. 2 deletion in individuals identified
through an affected relative: Cast a wide FISHing net! Genet Med.
3:23–29. 2001.
|
|
9
|
Cohen MM Jr, Gorlin RJ and Fraser FC:
Craniofacial disordersEmery and Rimoin's principles and practice of
medical genetics. Rimoin DL, et al: 3rd. Churchill Livingstone; New
York, NY: pp. 1121–1147. 1996
|
|
10
|
Gollo Dantas A, Bortolai A,
Moysés-Oliveira M, Takeno Herrero S, Azoubel Antunes A, Tavares
Costa-Carvalho B, Ayres Meloni V and Melaragno MI: 22q11.2 deletion
syndrome due to a translocation t(6;22) in a patient conceived via
in vitro fertilization. Mol Syndromol. 6:242–247. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jaquez M, Driscoll DA, Li M, Emanuel BS,
Hernandez I, Jaquez F, Lembert N, Ramirez J and Matalon R:
Unbalanced 15;22 translocation in a patient with manifestations of
DiGeorge and velocardiofacial syndrome. Am J Med Genet. 70:6–10.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Van Hove JL, McConkie-Rosell A, Chen YT,
Iafolla AK, Lanman JT Jr, Hennessy MD and Kahler SG: Unbalanced
translocation 46,XY,-15,+der(22)t(15;22)(q13;q11)pat: Case report
and review of the literature. Am J Med Genet. 44:24–30. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fryns JP, Kleczkowska A and van den Berghe
H: Frontonasal malformation and reciprocal translocation
t(15;22)(q22;q13). Clin Genet. 44:46–47. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pinto-Escalante D, Ceballos-Quintal JM,
Castillo-Zapata I and Canto-Herrera J: Full mosaic monosomy 22 in a
child with DiGeorge syndrome facial appearance. Am J Med Genet.
76:150–153. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Faed MJ, Robertson J, Beck JS, Cater JI,
Bose B and Madlom MM: Features of di George syndrome in a child
with 45,XX,-3,-22,+der(3),t(3;22)(p25;q11). J Med Genet.
24:225–227. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reddy KS, Sulcova V and Siassi B: Two sibs
with Wolf-Hirschhorn and DiGeorge deletions resulting from an
unbalanced chromosome rearrangement, 45,XX/XY, der(4)t(4;22)
(p16.3;q11.2) mat,-22. J Med Genet. 33:852–855. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dundar M, Kiraz A, Tasdemir S, Akalin H,
Kurtoglu S, Hafo F, Cine N and Savli H: Unbalanced 3;22
translocation with 22q11 and 3p deletion syndrome. Am J Med Genet
A. 152A:1–2795. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nur BG, Cetin Z, Clark OA, Mihci E, Oygur
N and Karauzum SB: 22q11.2 syndrome due to maternal translocation
t(18;22) (pl1.2;q11.2). Genet Couns. 26:67–75. 2015.PubMed/NCBI
|
|
19
|
McGoey RR and Lacassie Y: Paternal
balanced reciprocal translocation t(9;22)(q34.3;q11.2) resulting in
an infant with features of the 9q subtelomere and the 22q11
deletion syndromes due to 3:1 meiotic segregation and tertiary
monosomy. Am J Med Genet A. 149A:1–2542. 2009. View Article : Google Scholar
|
|
20
|
Kashevarova AA, Belyaeva EO, Nikonov AM,
Plotnikova OV, Skryabin NA, Nikitina TV, Vasilyev SA, Yakovleva YS,
Babushkina NP, Tolmacheva EN, et al: Compound phenotype in a girl
with r(22), concomitant microdeletion 22q13.32-q13.33 and mosaic
monosomy 22. Mol Cytogenet. 11:262018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sandrin-Garcia P, Macedo C, Martelli LR,
Ramos ES, Guion-Almeida ML, Richieri-Costa A and Passos GA:
Recurrent 22q11.2 deletion in a sibship suggestive of parental
germline mosaicism in velocardiofacial syndrome. Clin Genet.
61:380–383. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Adeyinka A, Stockero KJ, Flynn HC, Lorentz
CP, Ketterling RP and Jalal SM: Familial 22q11.2 deletions in
DiGeorge/velocardiofacial syndrome are predominantly smaller than
the commonly observed 3Mb. Genet Med. 6:517–520. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shprintzen RJ, Higgins AM, Antshel K,
Fremont W, Roizen N and Kates W: Velo-cardio-facial syndrome. Curr
Opin Pediatr. 17:725–730. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yagi H, Furutani Y, Hamada H, Sasaki T,
Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, et
al: Role of TBX1 in human del22q11.2 syndrome. Lancet.
362:1366–1373. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stoller JZ and Epstein JA: Identification
of a novel nuclear localization signal in Tbx1 that is deleted in
DiGeorge syndrome patients harboring the 1223delC mutation. Hum Mol
Genet. 14:885–892. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao S, Moreno M, Eliason S, Cao H, Li X,
Yu W, Bidlack FB, Margolis HC, Baldini A and Amendt BA: TBX1
protein interactions and microRNA-96-5p regulation controls cell
proliferation during craniofacial and dental development:
Implications for 22q11.2 deletion syndrome. Hum Mol Genet.
24:2330–2348. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bassett AS, Chow EW, Husted J, Weksberg R,
Caluseriu O, Webb GD and Gatzoulis MA: Clinical features of 78
adults with 22q11 deletion syndrome. Am J Med Genet A. 138:307–313.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oskarsdóttir S, Belfrage M, Sandstedt E,
Viggedal G and Uvebrant P: Disabilities and cognition in children
and adolescents with 22q11 deletion syndrome. Dev Med Child Neurol.
47:177–184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shprintzen RJ, Wang F, Goldberg R and
Marion R: The expanded velo-cardio-facial syndrome (VCF):
Additional features of the most common clefting syndrome. Am J Hum
Genet. 37:A771985.
|
|
30
|
Jurca A, Kinga K, Bembea M, Gug C and
Jurca C: Fanconi anemia with cleft palate. Rev Med Chir Soc Med Nat
Iasi. 118:1074–1077. 2014.PubMed/NCBI
|
|
31
|
Ryan AK, Goodship JA, Wilson DI, Philip N,
Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B,
Prieur M, et al: Spectrum of clinical features associated with
interstitial chromosome 22q11 deletions: A European collaborative
study. J Med Genet. 34:798–804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shprintzen RJ, Goldberg R, Young D and
Wolford L: The velo-cardiofacial syndrome: A clinical and genetics
analysis. Pediatr. 67:167–172. 1981.
|
|
33
|
Moss E, Wang P and McDonald-McGinn DM:
Characteristic cognitive profile in patients with a 22q11 deletion:
Verbal IQ exceeds nonverbal IQ. Am J Hum Genet. 57:A911995.
|
|
34
|
Wilson DI, Burn J, Scambler P and Goodship
J: DiGeorge syndrome: part of CATCH 22. J Med Genet. 30:852–856.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Capriotti TM and Parker Frizzell JP:
Pathophysiology. Introductory concepts and clinical perspectives.
1. F.A. Davis Company; Philadelphia, PA: 2016
|
|
36
|
Capra V, Mascelli S, Garrè ML, Nozza P,
Vaccari C, Bricco L, Sloan-Béna F, Gimelli S, Cuoco C, Gimelli G
and Tassano E: Parental imbalances involving chromosomes 15q and
22q may predispose to the formation of de novo pathogenic
microdeletions and microduplications in the offspring. PLoS One.
8:e579102013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Popovici C: Profilaxia bolilor
geneticeGenetică Medicală. Covic M, Ștefănescu D and Sandovici I:
2rd. Polirom, Iaşi; pp. 619–647. 2011
|