Introduction
Diabetic nephropathy (DN) is a leading cause of
chronic kidney disease (CKD) and a major complication of diabetes.
Hypertension, proteinuria and edema are frequent clinical features
of DN patients and experimental DN rats (1). Hypertension elevates blood pressure,
increases the kidney damages, and thus becomes an important reason
leading to diabetes, DN and end-stage renal failure (ESRF)
(1,2). In addition, the morbidity of
hypertension in diabetic patients was 1.5 ~ 3.0 times higher than
that in non-diabetic populations, showing mutual promotion effect
between hypertension and DN.
Diabetic rats always displayed increased oxygen
consumption and tissue hypoxia throughout the kidney (3). Chronic hypoxia causes several public
health problems including renal fibrosis, reduced renal function,
DN, hypertensive nephropathy, CKD, and is the final pathway to ESRF
(4–8). Chronic hypoxia-induced hypertension is
associated with microvascular and tubulointerstitial injury
(9,10). Chronic hypoxia also plays a central
pathogenic role in the process of end-stage renal disease (ESRD),
including ESRF (11).
The activation of hypoxia-inducible factors (HIFs),
however, could prevent the progression of DN (3). Hypoxia inducible factor-1α (HIF-1α) is
an oxygen-sensitive gene and is a transcription factor that
regulates a wide variety of genes (7). HIF-1α is recognized to be the essential
response regulator to hypoxia, and its expression has been
evidenced to be correlated with early progression of some diseases
including diabetic retinopathy which was caused by hypoxia-induced
hyperglycemia (6). Some evidences
show HIF-1α's activation was associated with hypertension of
artery, pulmonary or elsewhere (12–14).
Researches had shown that overexpression of HIF-1α in tubular
epithelial cells contributed to renal fibrosis progression, and
inhibition of HIF-1α expression prevented the progression of renal
fibrosis and attenuated DN progression (5,7,11,15).
They found HIF-1α-mediated abnormal pathways or genetic
dysregulations triggered hypertension (16). For instance, Luo et al showed
the elevated expression of endothelial HIF-1α in V-Cadherine/HIF-1α
floxed mice promoted the progression of angiotensin II
infusion-induced hypertensive CKD (5). On the contrary, the deletion of HIF-1α
was reported to attenuated chronic hypoxia induced hypertension,
proteinuria, kidney damage and so on (12–14,17).
Some studies showed stabilization or activation of HIF-1α was
essential under hypoxic conditions (7,18),
suggesting the important roles of HIF-1α expression in
hypertension. However, little is known about the roles of HIF-1α
deficiency in hypertensive DN progression.
The present study was designed to investigate the
effect of HIF-1α deficiency on DN with hypertension.
Mx/HIF-1α−/− mice were constructed and used as animal
HIF-1α−/−-deficient DN model in this study. Biochemical
and physiological changes in normal or HIF-1α deficient mice were
detected to assess the influence of HIF-1α deficiency on DN
progression.
Materials and methods
Animal and rats model
Mice labeled with Mx-Cre transgenic mice (JAXMICE:
003556, Strain name: B6.Cg-Tg (Mx1-Cre) 1Cgn/J) were mated to
HIF-1α gene LoxP mice (JAXMICE: 007561, B6.129-Hif1αtm3Rsjo/J,
HIF-1α homozygous floxed mice contain the HIF-1α exon 2 flanked by
LoxP sites), both were purchased from the Jackson Laboratories
(Jackson Laboratory, Ben Harbor, ME, USA). Mice were allowed twice
hybridization, and the identified Mx-Cre + and HIF-1α floxed mice
(Mx/HIF-1α−/−) in the third generation were used. Mice
were intraperitoneally injected with 400 µl/time poly (I:C;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) every 4 days for a
total of 3 times for interferon induction (19). DN induction were performed on C57BL/6
mice (12 weeks old, n=15) and Mx/HIF-1α−/− mice (n=15).
All mice received intraperitoneal injection of 140 mg/kg (weight
body) streptozotocin (STZ; Sigma-Aldrich; Merck KGaA). Another 15
mice received injection of equal volume normal saline and were used
as blank control. Mice were maintained on a normal diet. Diabetic
mice was recognized when fasting blood glucose (FBG) ≥16.7 mmol/l
at 72 h post STZ injection. Then animal body weight, 24 h urine
albumin, blood pressure, serum glucose were measured weekly.
Hypertensive model was recognized with elevated systolic blood
pressure (SBP) >140 mmHg. All animals were killed by
intraperitoneally administering overdose of sodium pentobarbital
(180 mg/kg body weight) at 16 weeks (20). These studies were approved by the
Institutional Animal Care and Use Committee Affiliated to Zhengzhou
University (Zhengzhou, China).
Blood pressure measurement
At the time of diabetic mice model construction,
mice SBP was measured once a week using noninvasive BP tail-cuff
with PowerLab system (ADInstruments, Mountain View, CA, USA) as
previously described (21,22). Prior to experimental protocols, mice
were trained for 2 weeks (twice a week). Moreover, acclimation
before each recording was needed.
Biochemistry investigation
Twenty-four hour urine were collected from housed
mice. Urine albumin was measured using a ELISA kit (Bethyl
Laboratories, Inc., Montgomery, TX, USA). FBG in tail vein were
detected weekly using a glucometer (OneTouch Ultra; Lifescan, Inc.,
Milpitas, CA, USA).
Histologic analysis
Sixteen weeks later, mice were killed and kidney
tissues were fixed, imbedded, and 2 µm sections were stained with
periodic acid-Schiff's (PAS) reagent at 37°C for 10 min and
hematoxylin at 37°C for 5 min, followed by image analysis using
Image Pro Plus (Media Cybernetics, Silver Spring, MD, USA).
Western blot analysis
Kidney tissues were lysed, prepared, and separated
using 10% SDS-PAGE. Then proteins were transblotted onto Millipore
PVDF membranes (EMD Millipore, Billerica, MA, USA) which were then
blocked with milk. Then membranes were subjected to incubation in
the specific primary antibody anti-HIF-1α (1:1,500 in dilution;
Cell Signaling Technology, Inc., Danvers, MA, USA) at 4°C
overnight, and HRP-conjugated secondary antibodies for 1 h. GAPDH
(1:2,000; Cell Signaling Technology, Inc.) was used as internal
reference gene. Immunoreactive protein bands were analyzed using a
Bio-Rad Quantity One software (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) by a chemiluminescence reaction system.
Statistical analysis
Data were recorded as means ± SD (standard
deviation). Analysis was performed using GraphPad Prism v.6.0
(GraphPad Software Inc., La Jolla, CA, USA). Statistical
differences among three groups were analyzed using one-way ANOVA
followed by Bonferroni's post hoc test. All datasets were
considered normally distributed and analyzed using parametric
statistics. P<0.05 was considered to indicate a statistically
significant difference.
Results
HIF-1α knockout promotes DN
pathogenesis
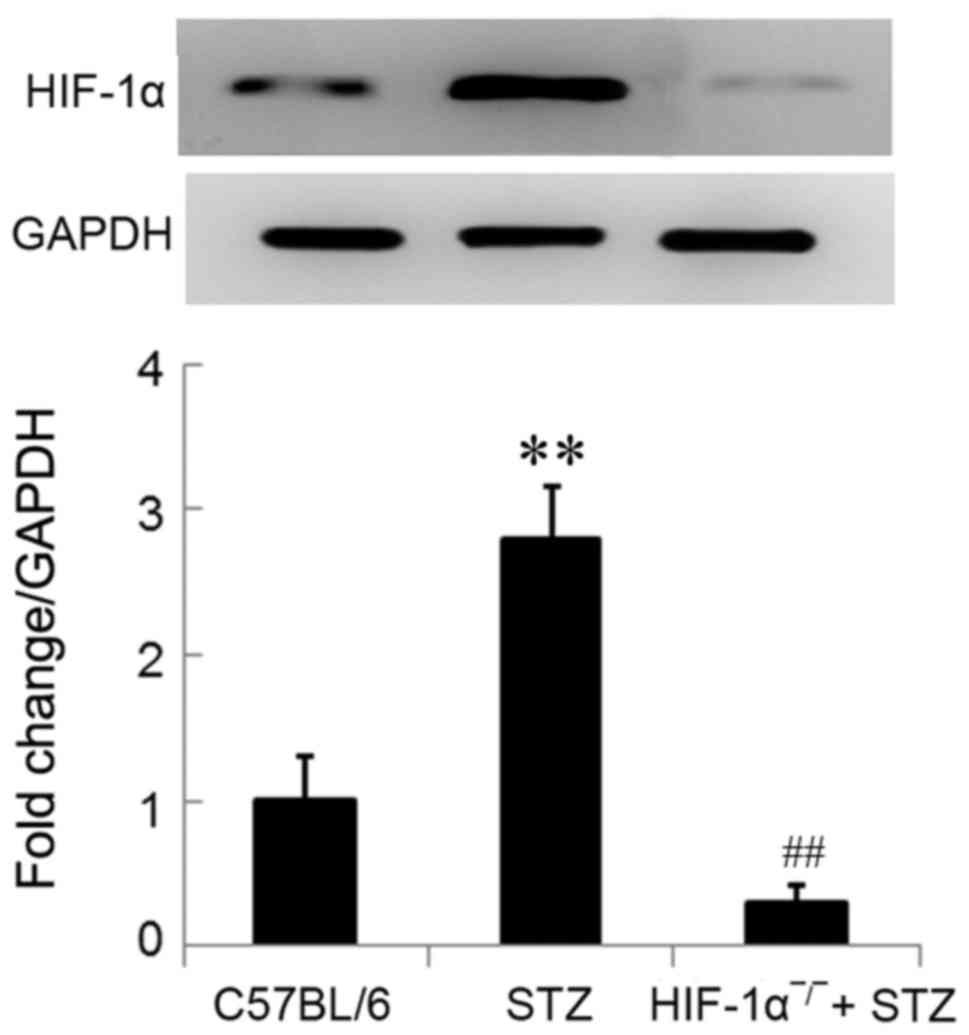
HIF-1α protein were significantly upregulated in
diabetic mice in comparison with the control mice (P<0.01;
Fig. 1). The significant
downregulation of HIF-1α protein (not complete knockout) in
Mx/HIF-1α−/− mice was determined using western blot
analysis, which confirmed the knockout of HIF-1α in
Mx/HIF-1α−/− mice (Fig.
1). The little expression of HIF-1α protein in
Mx/HIF-1α−/− mice might due to the HIF-1α homolog.
Effect of HIF-1α knockout on body
weight, urinary albumin, blood glucose and pressure
The body weight, FBG, 24 h urinary albumin, and SBP
in 3 groups were investigated. No death was observed in control
(C57BL/6) mice, and 5 mice in STZ group died during the 16 weeks.
All Mx/HIF-1α−/−mice died in the first 9 weeks. So the
investigation of HIF-1α−/−mice were available in the
first 9 weeks.
During the experimental protocol, non-diabetic mice
had the expected low level of FBG, urinary albumin, and SBP value.
In comparison, HIF-1α−/− + STZ mice showed the lowest
body weight, and the highest levels of FBG (>20 mmol/l), 24 h
urinary albumin and SBP during the 16 weeks' experimental protocol
(Fig. 2). As expected, the body
weight of non-diabetic mice was gradually increased during the
experiment period and was already significantly higher than mice
treated with STZ at the end of first week (P<0.01; Fig. 2A). On the contrary, the body weight
of mice with diabetes (STZ and HIF-1α−/− + STZ) was
declined during the experiment period. The FBG levels of the
diabetic mice (STZ and HIF-1α−/− + STZ) were
significantly higher than those of control mice (C57BL/6) at the
beginning (P<0.001), and were gradually decreased since the
third week, indicating the induction of diabetic mice by STZ
administration. In comparison with the gradually increased urinary
albumin level in HIF-1α−/− + STZ mice, that of the STZ
or C57BL/6 mice did not increased during all the experimental
protocol (Fig. 2C), revealing the
development of DN in HIF-1α−/− + STZ mice. No increment
in urinary albumin was observed in STZ-induced diabetic mice
compared with non-diabetic C57BL/6 mice (P>0.05), suggesting
there was no DN incidence in STZ-induced diabetic mice. At last we
found the diabetic mice displayed relative higher SBP values in
comparison with C57BL/6 mice (non-diabetic, P<0.01; Fig. 2D). The HIF-1α−/− + STZ
mice developed with SBP value of more than 140 mmHg at the fifth
week, indicating the development of hypertensive mice. The SBP
value in STZ group was lower than 140 mmHg, revealing there was no
hypertensive mice in STZ group although the SBP value was relative
higher than that in control mice (P<0.05). Taken together, these
results demonstrated that HIF-1α deficiency distinctly accelerated
the DN pathogenesis.
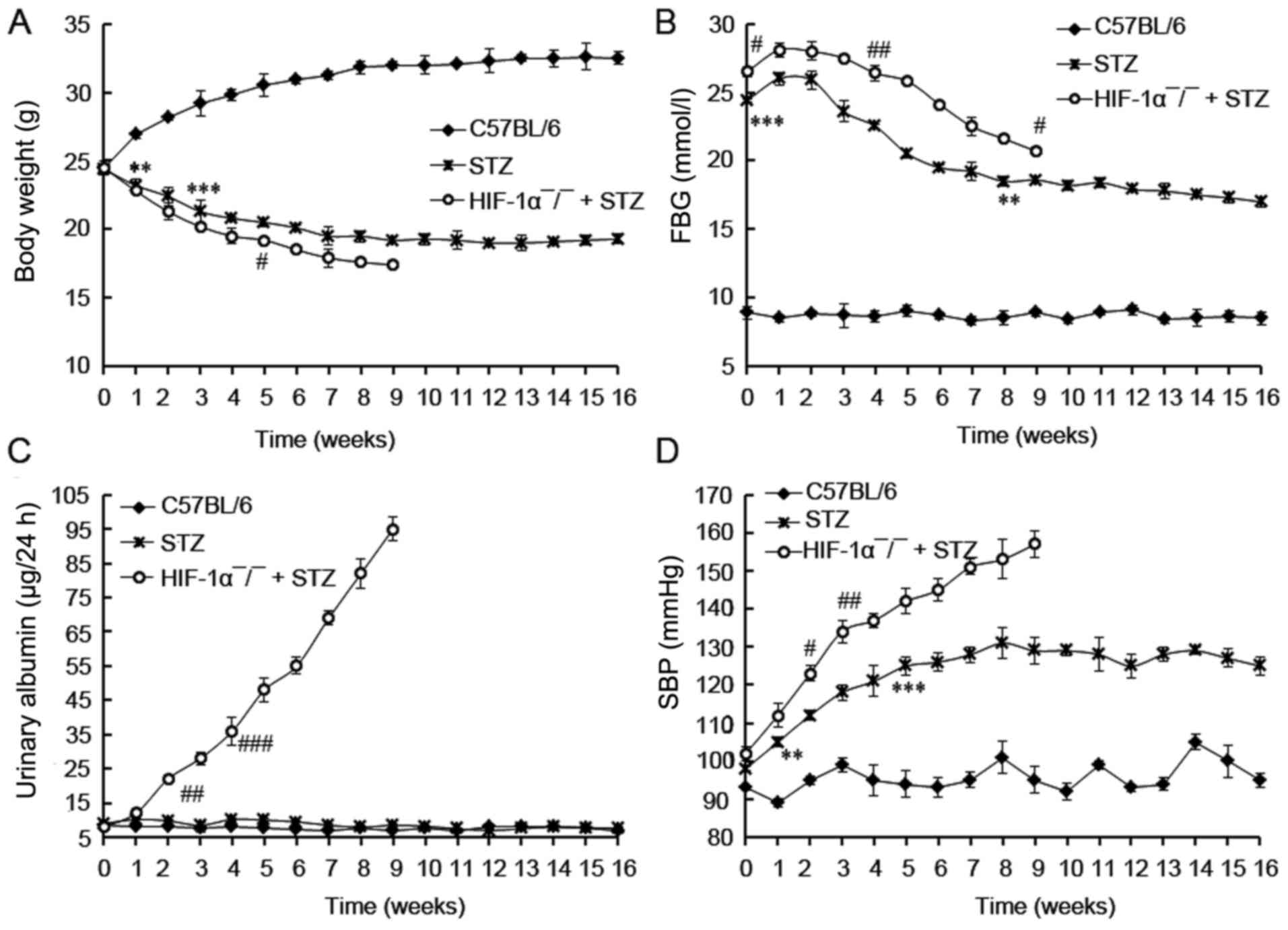 | Figure 2.The investigation of biochemical
parameters in diabetic rats. (A) Body weight; (B) FBG content; (C)
urinary albumin content; (D) BP content. Mice were treated with STZ
for induction of diabetes. The investigation of body weight, FBG,
urinary albumin, and SBP in mice were started at 3 days post STZ
performed weekly during 16 weeks post diabetic induction. HIF-1α
deficiency promotes DN progression. Data are expressed by mean ±
standard deviation (SD). ** and *** indicates differences at
P<0.01 and 0.001 between C57BL/6 and STZ treated mice. #, ##,
and ### indicates differences at P<0.05, 0.01, and 0.001 between
HIF-1α-/- + STZ and STZ treated mice. STZ,
streptozotocin; FBG, fasting blood glucose; SBP, systolic blood
pressure; DN, diabetic nephropathy. |
HIF-1α knockout promotes glomerular
damage
To determine the damage of HIF-1α deficiency on
kidney function, we performed the PAS staining on glomerular. We
found that diabetic mice with HIF-1α deficiency showed the
accelerated severity of glomerular damage compared with STZ-induced
diabetic mice (Fig. 3). Diabetic
mice, especially HIF-1α−/− mice, showed progressive
histopathologic changes in glomeruli and tubulointerstitium.
HIF-1α−/− mice showed distinct renal injury histologic
properties including collagen deposition in glomeruli, glomerular
sclerosis, necrotic lysis, tubular brush border loss, tubular
dilatation, and sloughing of cellular debris into the tubular
lumen. In comparison with HIF-1α−/− + STZ mice, mice in
STZ group showed slight histopathologic changes. These distinct
histopathologic changes in HIF-1α−/−diabetic mice
confirmed DN and suggested that HIF-1α deficiency promoted
glomerular damage.
Discussion
We confirmed that DN progression was associated with
downregulation of HIF-1α. We found the knockout of HIF-1α promoted
DN progression in mice. In comparison with STZ-induced diabetic
mice, HIF-1α deficient mice plus STZ showed increased FBG and SBP
level, and enhanced DN pathological characters of aggravated
glomeruli damage and elevated levels of 24 h urinary albumin. We
demonstrated that HIF-1α deficiency promoted the DN progression in
diabetic mice.
HIF-1α expression could be induced by hypoxia and
was correlated with early progression of some diseases (6). One of the leading causes of DN in
diabetes is renal hypoxia (23).
Elevated HIF-1α expression in kidney of mice with hypertensive
nephropathy and renal fibrosis had been proved to promote the
progression of diseases (5,11). On contrast, the deletion of HIF-1α
could attenuate disease progression, and showing protective effect
on kidney function. In the present study, we confirmed the
upregulation of HIF-1α in hypertensive DN kidneys of mice with
renal fibrosis. These demonstrated hypoxic conditions in DN
progression and the contribution to kidney damage.
Referring to the HIF-1α deficiency effect, reports
showed HIF-1α deficiency could attenuate hypertension of artery,
pulmonary, proteinuria, and kidney damage (12–14,17). As
reported, hypoxia-induced renal tubular epithelial cell death was
prevented by HIF-1α-deficiency, and that was a glucose dependent
manner (15). Luo et al
(5), demonstrated that HIF-1α
deficiency attenuated glomeruli damage and renal fibrosis in
angiotensin II-infused mice. Shimoda et al (17), showed an partial HIF-1α deficiency of
Hif1a (+/-) could blunt hypoxia-induced right ventricular
hypertrophy and polycythemia in vivo in PASMCs. Also, in
comparison with reduced K+ current density in PASMCs from HIF-1α
(+/+), no reduction in K+ current density was observed in PASMCs of
hypoxic HIF-1α (+/-) mice (17). Hu
et al (13), showed
overexpression of HIF-1α in fructose-induced salt-dependent
hypertensive rats attenuated high salt + fructose-induced high
blood pressure. Taken together, these results showed HIF-1α
deficiency benefited to attenuate hypertension or kidney
damage.
In the present study, however, we demonstrated that
HIF-1α deficiency accelerated hypertensive DN progression in
Mx/HIF-1α−/−mice. HIF-1α deficient mice showed
aggravated kidney damage, higher FBG and SBP levels, and elevated
collagen deposition, suggesting the progressive roles of HIF-1α
deficiency in STZ-induced DN. Our results was in accordance with
the that from Bohuslavova et al (24), who stated the heterozygosity HIF-1α
knockout (HIF-1α+/-) mice displayed diabetic cardiomyopathy with
decreased LVFS and myocardial function. They found STZ inhibited
HIF-1α expression in wild type mice and upregulated HIF-1α and Col1
in HIF-1α+/-mice, suggesting the genetic variation at the HIF-1α
locus influenced the extracellular matrix reorganization and risk
for fibrosis and diabetic cardiomyopathy. We guessed the aggravated
DN in HIF-1α-deficient mice might due to the HIF-1α
deficiency-induced deletion of compensatory effect to hypoxia
responses, that led to the loss of hypoxia preconditioning.
Finally, elevated hypoxic responses in kidney tissues promoted the
transdifferentiation of renal tubular epithelial cells to
fibroblasts, and facilitated renal fibrosis and irreversible kidney
damage.
In the present study, we also found that the urinary
albumin in HIF-1α deletion mice were significantly upregulated
compared with those in WT mice administrated with STZ. This
revealed that HIF-1α deficiency promoted damage on glomerular
filtration membrane, thus promoting the filtration of urinary
albumin to blood. However, no increment in urinary albumin was
found in STZ-treated WT mice compared with control. Generally, the
decreased body weight and increased FBG, urinary albumin and SBP
were predictors of DM (25). The
elevated urinary albumin is a hallmark of diabetic-related kidney
disease (26). We did not found the
upregulated urinary albumin in mice in WT group after being
administrated with STZ as HIF-1α deficient mice showed, but found
the slight histopathologic changes in kidney was in consistence
with the unchanged urinary albumin level. This fact might suggested
that STZ not definitely induced albumin increment, and also proved
that HIF-1α deficiency indeed promoted kidney damage.
Glucose inhibited hypoxia-induced HIF-1α
accumulation, and hyperglycemia regulated stability and function of
HIF-1α through proteasomal degradation in vivo (27). Gao et al (28), showed that hyperglycemia could
inhibit HIF-1α expression and attenuate the hypoxic-induced
apoptosis of bovine aortic smooth muscle cells (BASMC). So, they
suggested that hyperglycemia altered hypoxia-induced changes in
vascular cell growth by inhibiting HIF-1α. HIF-1α also mediates the
accumulation of collagen by activation of tissue inhibitor of
metalloproteinase-1 (TIMP-1) or TGF-β1/Smad3 signaling pathway. Fu
et al (29), showed HIF-1α
contributed to regulation of collagen accumulation and renal
fibrosis by cross talking with TGF-β1/Smad3 signaling pathway. To
further investigate the mechanism related to HIF-1α associated
hypertensive DN progression, we would perform more studies to
explore underlying mechanisms and signaling pathways.
In summary, we confirmed the HIF-1α upregulation in
DN kidney, together with the hyperglycemia and hypertensive. In
addition, HIF-1α deficiency did not attenuate DN damage, but
inversely accelerated DN progression. We speculated that there must
be differential pathways associated with HIF-1α deficiency-promoted
DN.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The data and materials used in this study are
available upon reasonable request from the corresponding author
once the paper has been published.
Authors' contributions
HuL and YJ participated in study design, data
analysis and manuscript drafting. HJ, HaL and YY screened patients,
collected and analyzed patient data, and revised the manuscript. YZ
and KZ were involved in experiment conduction and data
analysis.
Ethics approval and consent to
participate
The Ethical approval was obtained from the Ethics
Committee of The 1st Affiliated Hospital of Henan University of
Science and Technology, and all participants signed a consent form
prior to enrollment in the study.
Patient consent for publication
Not applicable.
Competing interests
All authors declare they have no competing
interests.
References
|
1
|
Xie S, Lu K, Zhang Y, Song X, Tan M and
Wang C: Effects of Jiangya Xiaoke prescription on TGF-β1 in
diabetic nephropathy rats with hypertension and its mechanisms. Int
J Clin Exp Med. 8:5129–5136. 2015.PubMed/NCBI
|
|
2
|
Grossman E: Should we treat
prehypertension in diabetes? What are the cons? Diabetes Care. 32
Suppl 2:S280–S283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nordquist L, Friederich-Persson M,
Fasching A, Liss P, Shoji K, Nangaku M, Hansell P and Palm F:
Activation of hypoxia-inducible factors prevents diabetic
nephropathy. J Am Soc Nephrol. 26:328–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gu HF, Zheng X, Abu Seman N, Gu T, Botusan
IR, Sunkari VG, Lokman EF, Brismar K and Catrina SB: Impact of the
hypoxia-inducible factor-1 α (HIF1A) Pro582Ser polymorphism on
diabetes nephropathy. Diabetes Care. 36:415–421. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo R, Zhang W, Zhao C, Zhang Y, Grenz A,
Eltzschig HK, Tou L, Kellems RW and Xia Y: Rt-pcr profiling reveals
the essential role of endothelial hif-1a in Hypertensive
Nephropathy. Hypertension. 62:A312013.
|
|
6
|
Yan H and Su Gf: Expression and
significance of HIF-1 α and VEGF in rats with diabetic retinopathy.
Asian Pac J Trop Med. 7:237–240. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kimura K, Iwano M, Higgins DF, Yamaguchi
Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, et
al: Stable expression of HIF-1alpha in tubular epithelial cells
promotes interstitial fibrosis. Am J Physiol Renal Physiol.
295:F1023–F1029. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nangaku M: Chronic hypoxia and
tubulointerstitial injury: A final common pathway to end-stage
renal failure. J Am Soc Nephrol. 17:17–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Steudel W, Scherrer-Crosbie M, Bloch KD,
Weimann J, Huang PL, Jones RC, Picard MH and Zapol WM: Sustained
pulmonary hypertension and right ventricular hypertrophy after
chronic hypoxia in mice with congenital deficiency of nitric oxide
synthase 3. J Clin Invest. 101:2468–2477. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mazzali M, Jefferson JA, Ni Z, Vaziri ND
and Johnson RJ: Microvascular and tubulointerstitial injury
associated with chronic hypoxia-induced hypertension. Kidney Int.
63:2088–2093. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fu W, Wang Y, Jin Z, Wang H, Cheng W, Zhou
H, Yin P and Peng W: Losartan alleviates renal fibrosis by
down-regulating HIF-1α and up-regulating MMP-9/TIMP-1 in rats with
5/6 nephrectomy. Ren Fail. 34:1297–1304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yamaji-Kegan K, Takimoto E, Semenza GL and
Johns RA: Hypoxia-inducible factor 1α is a critical downstream
mediator for hypoxia-induced mitogenic factor
(FIZZ1/RELMα)-mediated pulmonary hypertension, in A51. Experimental
models of pulmonary hypertension. Am J Respir Crit Care Med.
191:A19242015.
|
|
13
|
Hu J, Zhu Q, Li PL, Boini KM and Li N:
Inhibition of hypoxia inducible factor-1α in the renal medulla
contributes to fructose-induced salt-sensitive hypertension. FASEB
J. 30 1 Suppl:1216.22016.
|
|
14
|
Veith C, Zakrzewicz D, Dahal BK, Bálint Z,
Murmann K, Wygrecka M, Seeger W, Schermuly RT, Weissmann N and
Kwapiszewska G: Hypoxia-or PDGF-BB-dependent paxillin tyrosine
phosphorylation in pulmonary hypertension is reversed by HIF-1α
depletion or imatinib treatment. Thromb Haemost. 112:1288–1303.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Biju MP, Akai Y, Shrimanker N and Haase
VH: Protection of HIF-1-deficient primary renal tubular epithelial
cells from hypoxia-induced cell death is glucose dependent. Am J
Physiol Renal Physiol. 289:F1217–F1226. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bonnet S, Michelakis ED, Porter CJ,
Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil
R, McMurtry MS, et al: An abnormal mitochondrial-hypoxia inducible
factor-1alpha-Kv channel pathway disrupts oxygen sensing and
triggers pulmonary arterial hypertension in fawn hooded rats:
Uimilarities to human pulmonary arterial hypertension. Circulation.
113:2630–2641. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shimoda LA, Manalo DJ, Sham JS, Semenza GL
and Sylvester JT: Partial HIF-1alpha deficiency impairs pulmonary
arterial myocyte electrophysiological responses to hypoxia. Am J
Physiol Lung Cell Mol Physiol. 281:L202–L208. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Basu RK, Hubchak S, Hayashida T, Runyan
CE, Schumacker PT and Schnaper HW: Interdependence of HIF-1α and
TGF-β/Smad3 signaling in normoxic and hypoxic renal epithelial cell
collagen expression. Am J Physiol Renal Physiol. 300:F898–F905.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Du G, Leone M, Romeijn S, Kersten G,
Jiskoot W and Bouwstra JA: Immunogenicity of diphtheria toxoid and
poly(I:C) loaded cationic liposomes after hollow
microneedle-mediated intradermal injection in mice. Int J Pharm.
547:250–257. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu H, Li X, Yuan M, Wan W, Hu M, Wang X
and Jiang X: Intramyocardial delivery of bFGF with a biodegradable
and thermosensitive hydrogel improves angiogenesis and
cardio-protection in infarcted myocardium. Exp Ther Med.
14:3609–3615. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lim AK, Ma FY, Nikolic-Paterson DJ, Ozols
E, Young MJ, Bennett BL, Friedman GC and Tesch GH: Evaluation of
JNK blockade as an early intervention treatment for type 1 diabetic
nephropathy in hypertensive rats. Am J Nephrol. 34:337–346. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jara A, Benner CM, Sim D, Liu X, List EO,
Householder LA, Berryman DE and Kopchick JJ: Elevated systolic
blood pressure in male GH transgenic mice is age dependent.
Endocrinology. 155:975–986. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Franzén S: The role of hypoxia for the
development of diabetic nephropathy: Temporal relationship and
involvement of endothelin receptor signaling. Linköping Univ
Electron Press; pp. 532016
|
|
24
|
Bohuslavova R, Kolar F, Sedmera D,
Skvorova L, Papousek F, Neckar J and Pavlinkova G: Partial
deficiency of HIF-1α stimulates pathological cardiac changes in
streptozotocin-induced diabetic mice. BMC Endocr Disord. 14:112014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mori KP, Yokoi H, Kasahara M, Imamaki H,
Ishii A, Kuwabara T, Koga K, Kato Y, Toda N, Ohno S, et al:
Increase of total nephron albumin filtration and reabsorption in
diabetic nephropathy. J Am Soc Nephrol. 28:278–289. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Teumer A, Tin A, Sorice R, Gorski M, Yeo
NC, Chu AY, Li M, Li Y, Mijatovic V, Ko YA, et al: Genome-wide
association studies identify genetic loci associated with
albuminuria in diabetes. Diabetes. 65:803–817. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Catrina SB, Okamoto K, Pereira T, Brismar
K and Poellinger L: Hyperglycemia regulates hypoxia-inducible
factor-1alpha protein stability and function. Diabetes.
53:3226–3232. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao W, Ferguson G, Connell P, Walshe T,
Murphy R, Birney YA, O'Brien C and Cahill PA: High glucose
concentrations alter hypoxia-induced control of vascular smooth
muscle cell growth via a HIF-1alpha-dependent pathway. J Mol Cell
Cardiol. 42:609–619. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fu W, Wang Y, Jin Z, Wang H, Cheng W, Zhou
H, Yin P and Peng W: Losartan alleviates renal fibrosis by
down-regulating HIF-1α and up-regulating MMP-9/TIMP-1 in rats with
5/6 nephrectomy. Ren Fail. 34:1297–1304. 2012. View Article : Google Scholar : PubMed/NCBI
|