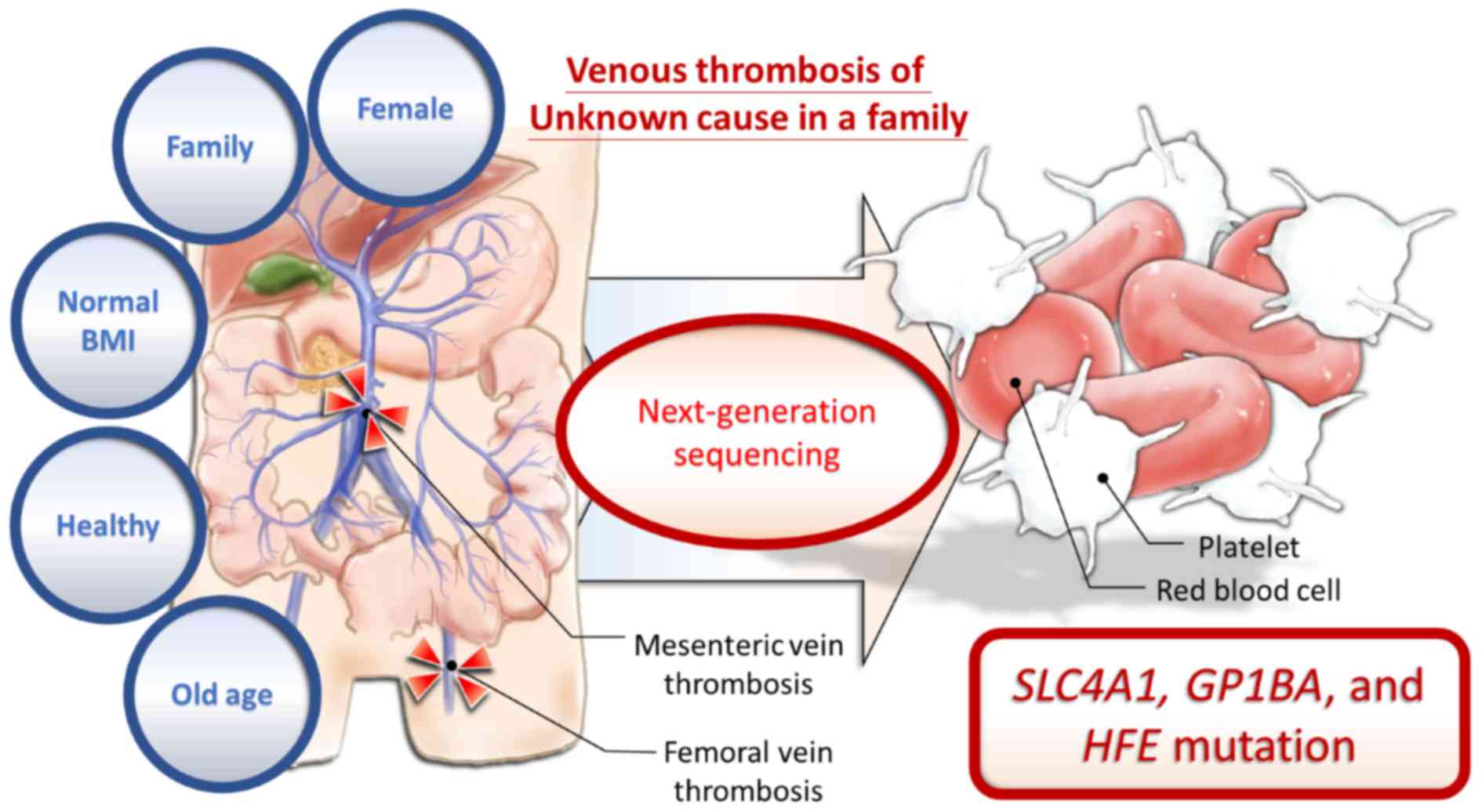
Introduction
Venous thrombosis is a common vascular event where a
blood clot forms within a vein. Age, surgery, obesity, trauma,
pregnancy and hormone replacement therapy are known risk factors of
venous thrombosis. Deficiencies of anti-thrombin, protein C and
protein S also pre-dispose to thrombosis. In addition, genetic
defects in fibrinogen, prothrombin, factor V Leiden, coagulation
factor XIII and coagulation factor XI are considered as inherited
risk factors (1–3). With the advance of sequencing
technology, rare and low-frequency genetic variants may now be
easily detected. Various novel genetic polymorphisms associated
with venous thrombosis, including deep venous thrombosis (DVT),
have been identified (4–6). Understanding of these rare inherited
risk factors may facilitate risk stratification and prophylaxis in
clinical practice. Venous thrombosis usually occurs in thrombosis
of veins in the leg and pulmonary embolism (1); however, venous thrombosis may also
occur in mesenteric veins and cause mesenteric venous thrombosis.
Primary mesenteric venous thrombosis is usually spontaneous or
idiopathic (7), accounting for
10–15% of all mesenteric ischemic events. Mesenteric ischemia
should be diagnosed and treated promptly, as the mortality rate may
reach 70% if the time to diagnosis is >24 h (8).
The present study presents the case of a
64-year-old, previously healthy Taiwanese woman admitted to
Kaohsiung Medical University Hospital (KMUH; Kaohsiung, Taiwan)
with mesenteric ischemia. The older sister of the proband also
suffered from DVT of the femoral vein at the same age. The younger
brother of the proband had intestinal arterial thrombosis when he
was 54 years of age. None of the patients had any specific
underlying diseases or risk factors for venous thrombosis;
therefore, it was hypothesized that familial genetic defects may
pre-dispose this pedigree to venous thrombosis. Peripheral blood
samples were collected from the proband, the proband's daughter and
the proband's older sister for next-generation sequencing (NGS).
Combined with bioinformatics, the present study aimed to identify
genetic factors associated with venous thrombosis in this
pedigree.
Materials and methods
Study subjects
A female patient (age, 64 years) was admitted to
KMUH (Kaohsiung, Taiwan) with mesenteric venous thrombosis of
unknown cause. The proband's older sister (thrombosis occurrence at
64 years; current age, 66 years) and younger brother (thrombosis
occurrence at 54 years; current age, 61 years) had also experienced
venous thrombotic events of unknown cause. The surveys for risk
factors of their thrombosis were all negative. In order to
investigate the possible cause of venous thrombosis in this
pedigree, peripheral blood samples of the proband, the proband's
older sister and the proband's daughter, a healthy 32-year-old
woman without any history of thrombotic events, were collected for
next-generation sequencing analysis. The present study was approved
by the Institutional Review Board of KMUH (Kaohsiung, Taiwan). All
subjects provided written informed consent.
Library preparation and
sequencing
For the generation of standard exome capture
libraries, the Agilent SureSelect XT Reagent kit for the Illumina
Hiseq paired-end sequencing library (cat. no. G9611A; Agilent
Technologies, Inc., Santa Clara, CA, USA) was used according to the
manufacturer's protocol. In all cases, the SureSelect XT Human All
Exon Version 6 (60 Mb) probe set (Agilent Technologies, Inc.) was
used. A total of 1 µg genomic DNA (gDNA) was used to construct a
library with the Agilent SureSelect XT Reagent kit. The
amplification adapter-ligated sample was purified using Agencourt
AMPure XP beads (Beckman Coulter, Brea, CA, USA) and analyzed on a
TapeStation 4200 D1000 screen tape (Agilent Technologies, Inc.).
The gDNA library (750 ng) was prepared for hybridization with the
capture beads, and the sample was hybridized for 24 h at 65°C,
captured with the Dynabeads MyOne Streptavidin T1 (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and purified using Agencourt
AMPure XP beads. The Agilent protocol was used for addition of
index tags (Agilent SureSelect XT Reagent kit; Illumina, Inc., San
Diego, CA, USA; cat. no. G9611B) by post-hybridization
amplification. Finally, all samples were sequenced on an Illumina
Sequencer (Illumina, Inc., San Diego, CA, USA) using the 150PE
protocol as specified by Illumina, Inc.
Processing of sequencing data and
identification of mutations
FASTQ file quality control was performed using
Trimmomatic (9). Reads from the
samples were mapped to the reference sequence Human Genome version
19 (hg19) by using the Burrows-Wheeler Aligner (10). For further variant analyses, variants
in coding or non-coding regions were identified, and annotation
were created with a Genome Analysis Toolkit (GATK) according to the
GATK best practices pipeline (11)
and Variant Effect Predictor (12).
These mutations were further filtered by the global minor allele
frequency and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) databases
(13). The mutations identified in
the ClinVar database were considered to be known variants and those
not found in the ClinVar database were identified as unknown
variants. Both known and unknown variants were further filtered out
by using the criterion >1% Asian minor allele frequency
according to TaiwanBioBank (http://taiwanview.twbiobank.org.tw) (14). The filtered variants were identified
as known or unknown rare variants.
Functional annotation analysis
Functional annotation was performed using the
Database for Annotation, Visualization and Integrated Discovery
(DAVID 6.7) (https://david.ncifcrf.gov) (15,16).
After annotating the gene list, the potential genes were obtained
through analysis of the genetic association database disease
class.
Protein features and domains
The information regarding features and functional
domains of protein sequences was adapted from UniProt [http://www.uniprot.org, P07359 (GP1BA_HUMAN)]. The
reference sequence of GP1BA from Homo sapiens (NCBI
Reference Sequence: NP_000164), Mus musculus (NCBI Reference
Sequence: NP_034456) and Rattus norvegicus (NCBI Reference
Sequence: NP_001103124) was obtained from the NCBI protein database
(https://www.ncbi.nlm.nih.gov/protein/).
Results and Discussion
Case presentation
The case of the present study was a 64-year-old
woman without any specific underlying diseases. She suffered from
progressive abdominal pain for 2 weeks and was brought to the
emergency department of KMUH, where abdominal computed tomography
revealed venous thrombosis involving the superior mesenteric vein
and splenic vein. Due to progressive abdominal pain with a
significant peritoneal sign, she underwent small bowel
segmentectomy and jejunostomy. After the operation, she received
anti-coagulant and anti-platelet therapies. The surveys for the
cause of her venous thrombosis, including examinations for cancer,
auto-immune diseases and coagulation diseases, were all negative
(Table I). Only elevated plasma
fibrinogen and D-dimer levels were identified; however, these are
common results during thrombotic events. The proband's older sister
had also suffered from DVT of the left femoral vein at the age of
64 years. She did not have any specific underlying diseases or
thrombosis risk factors, either. Furthermore, the proband's younger
brother had intestinal arterial thrombosis at 54 years of age.
Similarly, no specific underlying diseases or risk factors were
identified in this patient. These rare circumstances implied that
this pedigree may have familial genetic factors pre-disposing them
to thrombosis.
 | Table I.Biochemical parameters of the subjects
of the present study. |
Table I.
Biochemical parameters of the subjects
of the present study.
| Parameters | Normal range | Proband | Proband's
sister | Proband's
daughter |
|---|
| Age (years) |
| 64 | 66 | 34 |
| Gender |
| Female | Female | Female |
| BMI (kg/m2) |
| 24.4 | 22.6 | 17.5 |
| Rheumatoid factor
(mg/dl) | <15.9 | <10.0 | <10.0 | <10.0 |
| Lactate
(mmol/l) | 0.5–2.2 | 1.32 | <1.2 | <1.2 |
| Anti-thrombin III
(%) | 75–125 | 80 | 71 | 93 |
| Protein C (%) | 70–140 | 92 | 85 | 106 |
| Protein S (%) | 58.6–126 | 62 | 90 | 92 |
| Antinuclear
antibody | <1:40 | <1:40 | <1:40 | <1:40 |
| D-Dimer (mg/l) | <0.55 | 5.58a | 3.37a | 0.18 |
| Fibrinogen
(mg/dl) | 200–400 | 387.1 | 472.0a | 198.0 |
| PT (INR) | 0.85–1.15 | 1.07 | 1.02 | 1.05 |
| GLU (AC)
(mg/dl) | 65–109 | 150a | 114a | 96 |
| Albumin (g/dl) | 3.5–5 | 4.11 | 4.21 | 4.66 |
| LDH (IU/l) | 98–192 | 229a | 181 | 112 |
| BUN (mg/dl) | 8–20 | 8.6 | 9.9 | 9.6 |
| Creatinine
(mg/dl) | 0.44–1.03 | 0.52 | 0.76 | 0.59 |
| Na (mmol/l) | 136–144 | 140 | 136 | 141 |
| K (mmol/l) | 3.5–5.1 | 3.4 | 3.7 | 3.8 |
| CT (mg/dl) | 140–200 | 98 | 164 | 174 |
| TG (mg/dl) | 35–160 | 418 | 91 | 36 |
| APL IgG (GPL) | <15 | 7.46 | 11.21 | 4.31 |
| APL IgM (MPL) | <15 | <1.5 |
>100a | 2.17 |
| aCL-IgG
(GPL-U/ml) | <7 | 10 | 1.1 | 0.6 |
| aCL-IgM
(MPL-U/ml) | <10 | 4.3 |
>472a | 2.5 |
| WBC (×103/µl) | 4.14–10.52 | 11.41a | 6.98 | 5.83 |
| RBC (×106/µl) | 11.1–15.1 | 3.71 | 4.06 | 4.70 |
| Hgb (g/dl) | 11.1–15.1 | 10.8 | 12.6 | 13.6 |
| Hct (%) | 34.7–45.1 | 33.1a | 37.3 | 42.0 |
| MCV (fl) | 83.4–98.5 | 89.2 | 91.9 | 89.4 |
| PLT (×103/µl) | 160–370 | 201 | 213 | 269 |
The pedigree of the pedigree is presented in
Fig. 1. The proband (II-3) had
mesenteric vein thrombosis at 64 years of age. The proband's older
sister (II-1) had femoral vein thrombosis at 64 years of age also.
The proband's mother (I-2) had had a stroke. The proband's younger
brother (II-4) had intestinal arterial thrombosis. The laboratory
data of cases II-3 and II-1, as well as and the proband's daughter
(III-3), were collected and compared. Most of the data were within
normal ranges, except for plasma fibrinogen (Table I). The fibrinogen levels in case II-1
were slightly elevated, even though there was no active thrombotic
event when the blood sample was collected. Fibrinogen is an
important factor affecting platelet function (17). Furthermore, inherited fibrinogen
disorders are a risk factor of thrombotic complications (18). To investigate whether any genetic
variants on fibrinogen and other unknown genes were associated with
vein thrombosis within this pedigree, cases II-3, II-1 and III-3
were enrolled in the present study. Their peripheral blood samples
were collected for whole-exome sequencing.
Results of whole-exome sequencing
analysis: Known rare variants
After whole-exome sequencing, the results were
analyzed according to the flowchart presented in Fig. 2 and the procedures specified above.
Common variants were excluded from analysis. The rare variants were
further verified by ClinVar annotation based on hg19. Subsequently,
these genes were filtered out according to the criterion >1%
Asian minor allele frequency. Two genes with known rare variants
shared between the proband and the proband's sister are presented
in Table II. The first identical
mutation in the proband and the proband's sister was on solute
carrier family 4 member 1 (SLC4A1; GenBank ID NM_000342.3) which is
a band 3 cytoplasmic domain and phosphotransferase/anion
transporter (19). A heretogenous
missense variant c.388G>A (p.Gly130Arg, rs121912749) in exon 6
of SLC4A1 was observed. According to the ClinVar database, this
mutation of SLC4A1 is pathogenic in spherocytosis type 4.
Hereditary spherocytosis causes vein thrombosis in rare cases
(20,21). Although spherocytosis has not much in
common with venous thrombosis, the Gly130Arg mutation may still
lead to a decrease in anion exchange across the erythrocyte
membrane, thereby affecting the shape of the erythrocytes. Such a
shape change is expected to affect thrombosis.
| Table II.Gene mutations shared between the
proband and the proband's sister in ClinVar. |
Table II.
Gene mutations shared between the
proband and the proband's sister in ClinVar.
| Gene symbol | Nucleotide
change | Protein change | dbSNP no. |
|---|
| SLC4A1 | c.388G>A | p.Gly130Arg | rs121912749 |
| GP1BA |
c.1322_1344del23 |
p.Ser441TyrfsTer | rs770089708 |
The second gene with a rare mutation shared between
the proband and the proband's sister is glycoprotein Ib platelet α
subunit (GP1BA). GP1BA and GP1BB constitute a heterodimer of
glycoprotein Ib, which is a platelet surface membrane glycoprotein
(22). In platelets, GP1B is an
essential surface receptor for von Willebrand factor (VWF). The
binding site of VWF is located in the leucine-rich repeat domain of
GP1BA (23). The illustration of
protein domains, including the leucine-rich repeat N-terminal
domain [amino acids 19–46 (19–46 aa)], the NEL superfamily (34–205
aa) and the leucine-rich repeat C-terminal domain (221–281 aa) are
presented in Fig. 3. Mutations
within the leucine-rich repeat C-terminal domain of GP1BA
(p.Asp235Tyr, p.Trp246Leu or p.Met255Ile) may cause platelet-type
von Willebrand disease (24–26). The association between genetic
disorders of GP1BA and Bernard-Soulier syndrome (also known as
giant platelet syndrome), which is characterized by a low platelet
count and giant platelets, has been reported in Iranian, Indian and
Kuwaiti populations, even though the gene variants or frameshifts
are located in different regions of the GP1BA sequence (27–30).
Furthermore, a GP1BA polymorphism has been associated with
post-partum hemorrhage (31). In the
present study, the same deletion of the GP1BA gene
[c.1322_1344del23, p.Ser441TyrfsTer (frameshift and termination),
rs770089708] was detected in the proband and the proband's sister.
The VWF binding domain is located in the leucine-rich domain
(19–281 aa) of the extracellular region. The deletion was located
in the extracellular domain of GA1BA. The homology of in this
region of the GP1BA protein sequence is low among Homo
sapiens (NCBI Reference Sequence: NP_000164), Mus
musculus (NCBI Reference Sequence: NP_034456) and Rattus
norvegicus (NCBI Reference Sequence: NP_001103124). As the
deletion causes termination of GP1BA translation, the heterodimer
GP1BA/GP1BB on the platelet surface may be affected. In the ClinVar
database, the clinical significance of the GA1BP p.Ser441Tyr fs Ter
mutation was rated as ‘likely benign’, which was according to only
one data source. However, this mutation appears to be rare and may
still be involved in thrombosis. The mutation most likely leads to
the translation of a truncated GP1BA protein, which is soluble,
since it cannot be incorporated into the plasma membrane. A
detailed functional analysis of this GP1BA mutation requires to be
performed in the future.
Results of the whole exome analysis:
Unknown rare variants
The whole-exome analysis identified 742, 711 and 750
genes with unknown rare variants in cases II-3, II-1 and III-3,
respectively. A Venn diagram presenting shared genes with rare
mutations among the three subjects is displayed in Fig. 4. As cases II-3 and II-1 had presented
with venous thrombosis, the 168 identical variants were subjected
to a functional annotation analysis via DAVID bioinformatics
resources (14,15). The genetic association database
disease class for several of these gene mutations is presented in
Table III. Among them, 25 genes
were involved in human disease and 2 were associated with
hematological diseases. The first gene is homeostatic iron
regulator (HFE). The function of HFE protein is to regulate iron
absorption. Therefore, the defects in HFE result in iron storage
disorder and hereditary haemochromatosis (32). Previous studies have indicated an
association between thrombosis and hereditary hemochromatosis (with
HFE C282Y, H63D and S65C mutations) (33,34). In
the present study, cases II-3 and II-1 shared a HFE mutation,
namely an insertion in a splice region variant at 618–619/1519 of
HFE complementary DNA; this mutation has not been recorded in
ClinVar and has not been reported by any previous studies, to the
best of our knowledge. Although this variant did not alter the
coding sequence, this splice site mutation may alter the maturation
of HFE mRNA and then result in the production of abnormal proteins.
It was hypothesized that this novel HFE mutation may also be a risk
factor for venous thrombosis.
| Table III.Diseases associated with the shared
mutations between the proband and the proband's sister. |
Table III.
Diseases associated with the shared
mutations between the proband and the proband's sister.
| Gene symbol | Genetic association
database disease class |
|---|
| ATF5 | Psych |
| CALY | Psych |
| DOK1 | Cancer |
| EMX2 | Developmental,
reproduction |
| FADS1 | Immune, psych |
| GLRB | Neurological |
| HFEa | Aging, cancer,
cardiovascular, hematological, immune, infection, metabolic,
neurological, reproduction, vision |
| HLA-Ca | Cancer,
cardiovascular, hematological, immune, infection, metabolic,
neurological, normal variation, pharmacogenomic, reproduction |
| HLA-H | Immune |
| IL2RB | Cancer, immune,
infection, psych |
| KRT19 | Unknown (cirrhosis,
biliary primary) |
| LTBP4 | Immune |
| MASP1 | Immune |
| MMP10 | Cardiovascular |
| MTF1 | Neurological |
| MADCAM1 | Immune |
| MYO15A | Other |
| NOTCH4 | Immune,
neurological, psych |
| PANK2 | Neurological |
| PKHD1 | Renal |
| RBMX | Reproduction |
| RYK | Developmental |
| SCN1B | Neurological |
| SLC4A5 | Cardiovascular |
| ZBTB16 | Cancer |
The second shared gene associated with hematological
diseases was major histocompatibility complex, class I, C (HLA-C).
Two missense variants of HLA-C, namely c.98A>C (p.Asp33Ala,
rs1071650) and c.121C>A (p.Arg41Ser, rs41555420), were observed
in the two subjects via the Clinvar database. The function of these
were unknown; however, the Clinvar database did not record them as
pathogenic. As HLA-C is highly polymorphic and no published studies
have indicated that these variants of HLA-C are pathogenic, the
possibility of HLA-C being involved in abnormal venous thrombosis
was ruled out.
Results of whole exome analysis: All
rare variants among the proband, proband's sister and proband's
daughter
The identical mutations between cases II-3 and II-1
were discussed above, as these two subjects had presented with
venous thrombosis, while case III-3 was not considered, as she had
not experienced any thrombotic events. However, the possibility of
her developing venous thrombosis later in life could not be ruled
out. Therefore, the 313 identical variants among cases II-3, II-1
and III-3 were subjected to functional annotation analysis and the
genetic association database disease class of certain common
mutations is presented in Table IV.
Among them, only 5 genes, including HLA-A, HLA-B, major
histocompatibility complex, class II, DQβ1 (HLA-DQB1), HLA-DRB1 and
cytochrome P450 family 2 subfamily D member 6 (CYP2D6), were
associated with hematological diseases. HLA-A, HLA-B, HLA-DQB1 and
HLA-DRB1 are components of human major histocompatibility complex
class I and class II. Human major histocompatibility complex (MHC)
is a highly polymorphic gene, which recognises various pathogens.
Thus, it was assumed that these gene polymorphisms are not
associated with any abnormal venous thrombosis.
| Table IV.Diseases associated with the
mutations shared between the proband, the proband's sister and the
proband's daughter. |
Table IV.
Diseases associated with the
mutations shared between the proband, the proband's sister and the
proband's daughter.
| Gene symbol | Genetic association
database disease class |
|---|
| PRDM2 | Cancer |
| ALDH5A1 | Neurological,
psych |
| APOL4 | Psych, |
| ATR | Cancer,
cardiovascular |
| ATXN1 | Cardiovascular,
neurological, psych |
| ATXN3 | Neurological,
psych |
| ATN1 | Cardiovascular,
neurological, psych |
| CACNA1A | Neurological,
psych |
| CCL3L3 | Immune,
infection |
| CX3CR1 | Cancer,
cardiovascular, immune, infection, renal, vision |
| CYP2C19 | Cancer,
cardiovascular, immune, infection, metabolic, neurological, normal
variation, pharmacogenomic, psych, reproduction |
| CYP2D6a | Aging, cancer,
cardiovascular, chemdependency, hematological, immune, infection,
metabolic, neuro-logical, normal variation, pharmacogenomic, psych,
renal, reproduction, vision |
| DACH2 | Reproduction |
| DOCK3 | Psych |
| FAAH | Chemdependency,
metabolic, psych |
| FGFR1 | Developmental |
| FLG | Immune |
| FMO2 | Cancer |
| GPX1 | Cancer,
cardiovascular, immune, metabolic, pharmacogenomic, psych, renal,
vision |
| GNPAT | Psych |
| GDF5 | Metabolic |
| GHRHR | Cancer,
developmental, metabolic |
| IFI27 | Infection |
| KRT18 | Other |
| KIR3DL2 | Cancer, immune,
normal variation |
| KIR3DL3 | Cancer, normal
variation |
| KIR2DL1 | Cancer,
cardiovascular, immune, infection, normal variation, renal |
| LILRB1 | Immune |
| LILRB4 | Immune |
| HLA-Aa | Aging, cancer,
cardiovascular, hematological, immune, infection, metabolic,
neurological, normal variation, pharmacogenomic, psych,
reproduction |
| HLA-Ba | Cancer,
cardiovascular, hematological, immune, infection, metabolic,
neurological, normal variation, pharmacogenomic, reproduction |
| HLA-DQA1 | Cancer,
cardiovascular, developmental, immune, infection, metabolic, neuro
logical, normal variation, pharmacogenomic, psych, renal,
reproduction |
|
HLA-DQB1a | Cancer,
cardiovascular, hematological, immune, infection, metabolic, neuro
logical, normal variation, pharmacogenomic, psych, renal,
reproduction, vision |
|
HLA-DRB1a | Cancer,
cardiovascular, hematological, immune, infection, metabolic, neuro
logical, normal variation, pharmacogenomic, psych, renal,
reproduction, vision |
| HLA-DRB5 | Cancer,
cardiovascular, immune, infection, metabolic, neurological |
| MAP3K1 | Cancer |
| MUC2 | Immune |
| MUC3A | Immune |
| MUC4 | Immune,
reproduction |
| MUC5AC | Immune |
| MUC6 | Infection |
| MSH6 | Cancer |
| MYOC | Vision |
| NIPA1 | Immune |
| PER3 | Aging,
chemdependency, neurological, psych |
| RAI1 | Smith-magenis
syndrome |
| SLC5A4 | Psych |
| SYN2 | Psych |
| TNS1 | Developmental |
| TCN2 | Cancer,
cardiovascular, developmental, immune, infection, metabolic, neuro
logical, reproduction |
| TTN | Cardiomyopathy
dilated 1G, cardiomyopathy familial hypertrophic, 9, muscular
dystrophy limb-girdle type 2J, myopathy early-onset with fatal
cardiomyopathy, myopathy, proximal with early respiratory muscle
involvement, tibial muscular dystrophy tardive |
| XYLT2 | Immune, renal |
CYP2D6 is an important hepatic enzyme involved in
the metabolism of 20–25% of clinical drugs (35). CYP2D6 is highly polymorphic and
>100 allelic variants of CYP2D6 have been identified (36), several of which do not alter the
enzyme function of CYP2D6, although these known CYP2D6
polymorphisms may interfere with alternative splicing and
transcription of the mRNA (37). In
addition, CYP2D6 has a frequency of multiplication of 45% in Asian
populations (38). In the present
study, a homologous CYP2D6 variant c.1457G>C (p.Ser486Thr,
rs1135840) was detected among all three subjects. As the copy
number was not determined in the present study, it was not possible
to determine whether the subjects carried a CYP2D6 multiplication.
According to a previous study, the polymorphisms in CYP2D6*2 with
rs1135840 are common in Chinese populations and CYP2D6*2 with
rs1135840 retains its normal enzyme function (39). This may imply that the homologous
CYP2D6 variant does not affect the enzyme function in this
pedigree. Based on the above evidence, the identical gene mutations
among the three subjects of the present study may not be major
causes of venous thrombosis.
In conclusion, the present study identified common
SLC4A1 and GP1BA mutations, which are known in ClinVar, and the
common unknown HFE mutation between two female siblings with venous
thrombosis (Fig 5). To the best of
our knowledge, the present study was the first to report a possible
association between venous thrombosis and these mutations in an
Asian pedigree.
Acknowledgements
The authors would like to thank the Center for
Research Resources and Development of Kaohsiung Medical
University.
Funding
The present study was supported by grants from the
Ministry of Science and Technology (grant nos. MOST
106-2314-B-037-016-MY2, MOST 106-2320-B-037-029-MY3 and MOST
107-2320-B-037-011-MY3), Kaohsiung Medical University Hospital
(grant nos. KMUHS10701, KMUHS10712, KMUH106-6R15 and KMUH106-6R77),
and the Kaohsiung Medical University (grant no. KMU-DK108003).
Availability of data and materials
The analyzed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WC, MY and PK designed the study; WC and CS enrolled
the patients; WC, KL, JS, MY and PK analyzed the data and
interpreted the results; and WC, and MY wrote the manuscript. The
final version of the manuscript has been read and approved by all
authors, and each author believes that the manuscript represents
honest work.
Ethical approval and consent to
participate
The present study was approved by the Institutional
Review Board of KMUH (Kaohsiung, Taiwan). All subjects provided
written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rosendaal FR and Reitsma PH: Genetics of
venous thrombosis. J Thromb Haemost. 7 Suppl 1:S301–S304. 2009.
View Article : Google Scholar
|
|
2
|
Miyata T, Maruyama K, Banno F and Neki R:
Thrombophilia in East Asian countries: Are there any genetic
differences in these countries? Thromb J. 14 Suppl 1:S252016.
View Article : Google Scholar
|
|
3
|
Makris M, Rosendaal FR and Preston FE:
Familial thrombophilia: Genetic risk factors and management. J
Intern Med Suppl. 740:9–15. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lotta LA, Wang M, Yu J, Martinelli I, Yu
F, Passamonti SM, Consonni D, Pappalardo E, Menegatti M, Scherer
SE, et al: Identification of genetic risk variants for deep vein
thrombosis by multiplexed next-generation sequencing of 186
hemostatic/pro-inflammatory genes. BMC Med Genomics. 5:72012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lotta LA, Tuana G, Yu J, Martinelli I,
Wang M, Yu F, Passamonti SM, Pappalardo E, Valsecchi C, Scherer SE,
et al: Next-generation sequencing study finds an excess of rare,
coding single-nucleotide variants of ADAMTS13 in patients with deep
vein thrombosis. J Thromb Haemost. 11:1228–1239. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morange PE, Suchon P and Trégouët DA:
Genetics of venous thrombosis: Update in 2015. Thromb Haemost.
114:910–919. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sulger E and Gonzalez L: Mesenteric venous
thrombosisStatPearls [Internet]. Treasure Island (FL): StatPearls
Publishing; 2017, https://www.ncbi.nlm.nih.gov/books/NBK459184/January
15–2018
|
|
8
|
Singh M, Long B and Koyfman A: Mesenteric
ischemia: A deadly miss. Emerg Med Clin North Am. 35:879–888. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
McLaren W, Gil L, Hunt SE, Riat HS,
Ritchie GR, Thormann A, Flicek P and Cunningham F: The ensembl
variant effect predictor. Genome Biol. 17:1222016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Landrum MJ, Lee JM, Benson M, Brown G,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al:
ClinVar: Public archive of interpretations of clinically relevant
variants. Nucleic Acids Res. 44:D862–D868. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen CH, Yang JH, Chiang CWK, Hsiung CN,
Wu PE, Chang LC, Chu HW, Chang J, Song IW, Yang SL, et al:
Population structure of Han Chinese in the modern Taiwanese
population based on 10,000 participants in the Taiwan Biobank
project. Hum Mol Genet. 25:5321–5331. 2016.PubMed/NCBI
|
|
15
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
da Huang W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kunicki TJ, Williams SA and Nugent DJ:
Genetic variants that affect platelet function. Curr Opin Hematol.
19:371–379. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Korte W, Poon MC, Iorio A and Makris M:
Thrombosis in inherited fibrinogen disorders. Transfus Med
Hemother. 44:70–76. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Y, Yang J and Chen LM: Structure and
function of SLC4 family [formula: See text] transporters. Front
Physiol. 6:3552015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perkins LA, Jones SF and Bhargava RS:
Dural venous thrombosis following splenectomy in a patient with
hereditary spherocytosis. South Med J. 102:542–545. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Agarwal SK, Binbrek AS, Thompson JA and
Siddiqui SA: Massive pulmonary embolism and acute limb ischaemia in
a patient of hereditary spherocytosis and patent foramen ovale.
Heart Lung Circ. 19:742–744. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ozaki Y, Asazuma N, Suzuki-Inoue K and
Berndt MC: Platelet GPIb-IX-V-dependent signaling. J Thromb
Haemost. 3:1745–1751. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Watkins NA, Gusnanto A, de Bono B, De S,
Miranda-Saavedra D, Hardie DL, Angenent WG, Attwood AP, Ellis PD,
Erber W, et al: A HaemAtlas: Characterizing gene expression in
differentiated human blood cells. Blood. 113:e1–e9. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Enayat S, Ravanbod S, Rassoulzadegan M,
Jazebi M, Tarighat S, Ala F, Emsley J and Othman M: A novel D235Y
mutation in the GP1BA gene enhances platelet interaction with von
Willebrand factor in an Iranian family with platelet-type von
Willebrand disease. Thromb Haemost. 108:946–954. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lavenu-Bombled C, Guitton C, Dupuis A,
Baas MJ, Desconclois C, Dreyfus M, Li R, Caron C, Gachet C,
Fressinaud E and Lanza F: A novel platelet-type von Willebrand
disease mutation (GP1BA p. Met255Ile) associated with type 2B
‘Malmö/New York’ von Willebrand disease. Thromb Haemost.
116:1070–1078. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Woods AI, Sanchez-Luceros A, Bermejo E,
Paiva J, Alberto MF, Grosso SH, Kempfer AC and Lazzari MA:
Identification of p.W246L as a novel mutation in the GP1BA gene
responsible for platelet-type von Willebrand disease. Semin Thromb
Hemost. 40:151–160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ali S, Ghosh K and Shetty S: Novel genetic
abnormalities in bernard-soulier syndrome in india. Ann Hematol.
93:381–384. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shlebak A, Poles A, Manning R, Almuhareb
S, De La Funte J, Mitchell M and Lucas G: A novel homozygous
c.800C>G substitution in GP1BA exon 2 in a kuwaiti family with
bernard-soulier syndrome. Acta Haematol. 134:193–198. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Afrasiabi A, Lecchi A, Artoni A, Karimi M,
Ashouri E, Peyvandi F and Mannucci PM: Genetic characterization of
patients with bernard-soulier syndrome and their relatives from
Southern Iran. Platelets. 18:409–413. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ali S, Shetty S and Ghosh K: A novel
mutation in GP1BA gene leads to mono-allelic bernard soulier
syndrome form of macrothrombocytopenia. Blood Coagul Fibrinolysis.
28:94–95. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Biguzzi E, Franchi F, Acaia B, Ossola W,
Nava U, Paraboschi EM, Asselta R and Peyvandi F: Genetic background
and risk of postpartum haemorrhage: Results from an Italian cohort
of 3219 women. Haemophilia. 20:e377–e383. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Valenti L and Pelusi S: HFE mutations and
iron in hemodialysis patients. Hemodial Int. 21 Suppl 1:S47–S57.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brown K, Luddington R, Taylor SA,
Lillicrap DP and Baglin TP: Risk of venous thromboembolism
associated with the common hereditary haemochromatosis Hfe gene
(C282Y) mutation. Br J Haematol. 105:95–97. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dionisio Tavares Niewiadonski V, Dos
Santos Bianchi JV, de Almeida-Neto C, Gaburo N Jr and Sabino EC:
Evaluation of a high throughput method for the detection of
mutations associated with thrombosis and hereditary hemochromatosis
in Brazilian blood donors. PLoS One. 10:e01254602015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ingelman-Sundberg M: Pharmacogenetics of
cytochrome P450 and its applications in drug therapy: The past,
present and future. Trends Pharmacol Sci. 25:193–200. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang B, Yang LP, Zhang XZ, Huang SQ,
Bartlam M and Zhou SF: New insights into the structural
characteristics and functional relevance of the human cytochrome
P450 2D6 enzyme. Drug Metab Rev. 41:573–643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang D, Poi MJ, Sun X, Gaedigk A, Leeder
JS and Sadee W: Common CYP2D6 polymorphisms affecting alternative
splicing and transcription: Long-range haplotypes with two
regulatory variants modulate CYP2D6 activity. Hum Mol Genet.
23:268–278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sistonen J, Fuselli S, Levo A and
Sajantila A: CYP2D6 genotyping by a multiplex primer extension
reaction. Clin Chem. 51:1291–1295. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Goh LL, Lim CW, Sim WC, Toh LX and Leong
KP: Analysis of genetic variation in CYP450 genes for clinical
implementation. PLoS One. 12:e01692332017. View Article : Google Scholar : PubMed/NCBI
|