Introduction
Acute lung injury (ALI) or acute respiratory
distress syndrome (ARDS), as a clinically common lethal disease, is
characterized by extensive neutrophilic influx into the lungs, the
expression of pro-inflammatory mediators and damage of the lung
epithelium and endothelium, which result in pulmonary edema and the
deterioration of gas exchange (1,2). The
pathogenesis of ALI is considered to involve lung inflammation and
cell apoptosis, characterized by the accumulation of inflammatory
cells, the aberrant release of proteases, reactive oxygen species
(ROS) and proinflammatory cytokines (3,4).
Lipopolysaccharide (LPS) of gram-negative bacteria has been
suggested to be an important etiological factor responsible for
lung diseases, characterized by the presence of apoptosis in the
endothelium (5). Apoptosis of
pulmonary microvascular endothelial cells (PMVECs) damages the
barrier function of the pulmonary microvascular endothelium. Thus,
inhibition of the apoptosis of PMVECs is a crucial intervention
measure to prevent the occurrence of ALI (6,7).
Angiopoietins (Ang) are Tie2 receptor ligands that
play important roles in vascular development, vessel remodeling and
angiogenesis (8). Ang-1 and Ang-2
are the most specific ligands of Tie2, and Ang-2 has been shown to
be a competitive antagonist for Ang-1 at the receptor tyrosine
kinase Tie2 in endothelial cells (9). Ang-2, a secreted oligomeric
glycoprotein, stimulates endothelial cells and increases vascular
inflammation (10). In vitro
experiments have confirmed that Ang-2, under certain circumstances,
induces the phosphorylation of Tie-2 receptors, protein kinase B
(also known as Akt), extracellular signal-related kinase (ERK)1/2
and p38 members of the mitogen-activated protein kinase family
(11). In vivo,
Ang-2-deficient mice did not exhibit any vascular inflammatory
responses in LPS-induced sepsis experiments (12). Exceptionally high levels of
circulating Ang-2 have been observed in human sepsis and correlated
with mortality (10).
In addition, Harfouche et al (13) demonstrated that ERK1/2 and p38 are
activated by Ang-1 in endothelial cells. Harfouche and Hussain
(11) also reported that Ang-2
evokes p38 phosphorylation as strongly as that elicited by Ang-1.
The finding that Ang-2 promotes endothelial cell survival through
the phosphoinositide 3-kinase (PI3K) and ERK1/2 pathways suggests
that Ang-2 exerts qualitatively similar anti-apoptotic effects to
those elicited by Ang-1 (13,14).
However, little is known about the possible impact
of Ang-2 mediation on the LPS-induced apoptosis of rat PMVECs
(RPMVECs) and the mechanisms by which the mitogen-activated protein
kinase (MAPK) signaling pathway contributes to lung injury.
Therefore, in the present study, the LPS-induced expression of
Ang-2 was examined and the role of the p38 and ERK1/2 signaling
pathway in the apoptotic damage of PMVECs was investigated. The
study also focused on the possible influence of MAPK inhibitors on
PMVEC apoptosis. Moreover, p38 and ERK1/2 signaling pathways were
examined to elucidate whether Ang-2 acts as an upstream factor of
MAPK pathways in the modulation of the LPS-induced apoptosis of
PMVECs.
Materials and methods
Materials
RPMVECs were obtained from Shanghai Biomart
Technology Co., Ltd. (Shanghai, China). Dulbecco's modified Eagle's
medium (DMEM) and fetal bovine serum (FBS) were purchased from
Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA),
Antibodies to p38 (no. 14451), p-p38 (no. 4511), ERK1/2 (no. 4695),
p-ERK1/2 (no. 8544), Bcl-2 (no. 15071) and Bax (no. 5023) were
obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA).
Ang-2 antibody (no. ab155106) was purchased from Abcam (Shanghai,
China). β-actin antibody (no. sc-2357 and sc-2005), SB203580 and
PD98059 were obtained from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA). Recombinant human Ang-2 was purchased from PeproTech,
Inc. (Rocky Hill, NJ, USA). LPS (no. sc-3535) was obtained from
Santa Cruz Biotechnology, Inc. The Annexin V-fluorescein
isothiocyanate (FITC) kit was acquired from BestBio Co. (Shanghai,
China). TRIzol reagent was purchased from Invitrogen (Thermo Fisher
Scientific, Inc.), and the RevertAid First Strand cDNA Synthesis
kit and Maxima SYBR Green/ROX qPCR Master Mix were obtained from
Thermo Fisher Scientific, Inc.
Cell culture and treatment
RPMVECs were cultured in DMEM with 10% FBS, 1%
penicillin-streptomycin (Lonza, Cologne, Germany) and 1.5 g/l
glucose at 37°C in a humidified atmosphere containing 5%
CO2. Cells from passages 5–8 were used for all
experiments, and harvested with trypsin (0.25%) and EDTA (0.03%)
when the cells had reached exponential growth. RPMVECs were
incubated with LPS (10 µg/ml) and Ang-2 (300 ng/ml) for 6, 12, 24 h
respectively, or were incubated with Ang-2-alone, Ang-2 plus
SB203580 (300 ng/ml + 10 µM), and Ang-2 plus PD98059 (300 ng/ml +
30 µM) for 24 h prior to analysis by western blot, flow cytometry
and RT-qPCR.
Observation and detection of apoptosis
by fluorescence microscopy and flow cytometry
Cells were harvested and the percentage of apoptosis
was measured by flow cytometry using an Annexin V-FITC kit
according to the manufacturer's instructions. Following the
treatment, the adherent and floating cells were collected, washed
twice with cold PBS (4°C) and resuspended in 400 µl binding buffer.
Cells were first incubated with 5 µl Annexin V-FITC at room
temperature in the dark for 15 min and then with 10 µl propidium
iodide (PI; 40 µg/ml) at room temperature in the dark for 5 min.
Cells were observed under fluorescence microscopy and cell
suspensions were transferred to test tubes and detected by flow
cytometry. Cells with no drug treatment were used as a control.
Data were analyzed using CellQuest software version 3.3 (BD
Biosciences, San Jose, CA, USA).
Western blot analysis
Cells were washed with ice-cold PBS solution twice
and incubated for 1 h at 4°C in lysis buffer comprised of the
following: 150 mM NaCl, 50 mmol/l Tris HCl pH 8.0, 1% TritonX-100,
100 µg/ml phenylmethane sulfonyl fluoride. The lysates were
centrifuged at 13,380 × g for 30 min at 4°C and the supernatant was
collected. A total of 10 µG protein was loaded in each lane,
separated by 10% SDS-PAGE and transferred to PVDF membranes (EMD
Millipore, Billerica, MA, USA) by electroblotting for 2 h at 100 V.
Membranes were blocked for 1 h in 5% non-fat milk in PBS and then
incubated with primary antibodies (Ang-2, p38, p-p38, ERK1/2,
p-ERK1/2, Bcl-2 and Bax; 1:1,000 dilution) at 4°C overnight. The
membrane was washed with PBS solution and then incubated with
peroxidase-conjugated anti-rabbit (no. sc-2357; Santa Cruz
Biotechnology, Inc.) or anti-mouse (no. sc-2005; Santa Cruz
Biotechnology, Inc.) secondary antibody (1:4,000 dilution) for 1 h
at room temperature. The blots were assayed by chemiluminescence
(EMD Millipore) on X-ray film. Finally, the bands were analyzed
using ImageJ 1.43 software (National Institutes of Health,
Bethesda, MA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the RPMVECs using
TRIzol reagent. Complementary DNA (cDNA) was synthesized from 2 µg
total RNA and 1 µl oligo (dT), diluted to a volume of 8 µl in
DEPC-treated water, and heated at 70°C for 5 min. Then, 4 µl 5X
Reverse Transcription buffer, 2 µl Superscript reverse
transcriptase, 2 µl 2.5 mM dNTP mix, 1 µl RNase inhibitor and
DEPC-treated water were added to 20 µl. The mixture was gently
incubated at 37°C for 5 min, incubated at 42°C for 60 min to
synthesize cDNA and then heated at 70°C for 10 min to stop cDNA
synthesis. The cDNA was stored at −20°C. qPCR was performed using
the SYBR Green qPCR Master mix in the ABI 7300 Real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The reaction
volume was 20 µl, and the reaction mixture comprised 10 µl
SYBR-Green qPCR Master mix, 1 µl cDNA, 0.5 µl each of sense and
antisense primers, and water to 20 µl. The amplification profile
was as follows: Initial denaturation at 95°C for 10 min, followed
by 45 cycles at 95°C for 15 sec and 60°C for 30 sec. Primers for
Ang-2, Bcl-2, Bax and β-actin were designed with Primer Premier 5.0
software for the rat (Invitrogen; Thermo Fisher Scientific, Inc.)
as follows: Ang-2, sense 5′-CTGAAGATCCAGCTGAAG-3′ and antisense
5′-ATTGTCCGAATCCTTTGT-3′; Bcl-2, sense 5′-ATGTGTGTGGAGAGCGTCAACC-3′
and antisense 5′-CCAGGAGAAATCAAACAGAGGC-3′; Bax, sense
5′-GCGATGAACTGGACAACAACAT-3′ and antisense
5′-TAGCAAAGTAGAAAAGGGCAACC-3′; β-actin, sense
5′-TCATGAAGTGTGACGTTGACATCCGT-3′ and antisense
5′-CCTAGAAGCATTTGCGGTGCAGGATG-3′. Relative gene expression data
were calculated from the formula: 2−ΔΔCq, where Cq
represents the fractional cycle number at which the amount of
target reaches a fixed threshold (15).
Statistical analysis
All data are expressed as mean ± standard deviation.
Data were analyzed by repeated measures, one-way analysis of
variance. P<0.05 was considered to indicate a statistically
significant difference. Data were analyzed using the commercially
available SPSS software package, version 18.0 (SPSS, Inc., Chicago,
IL, USA). All data were obtained from three separate
experiments.
Results
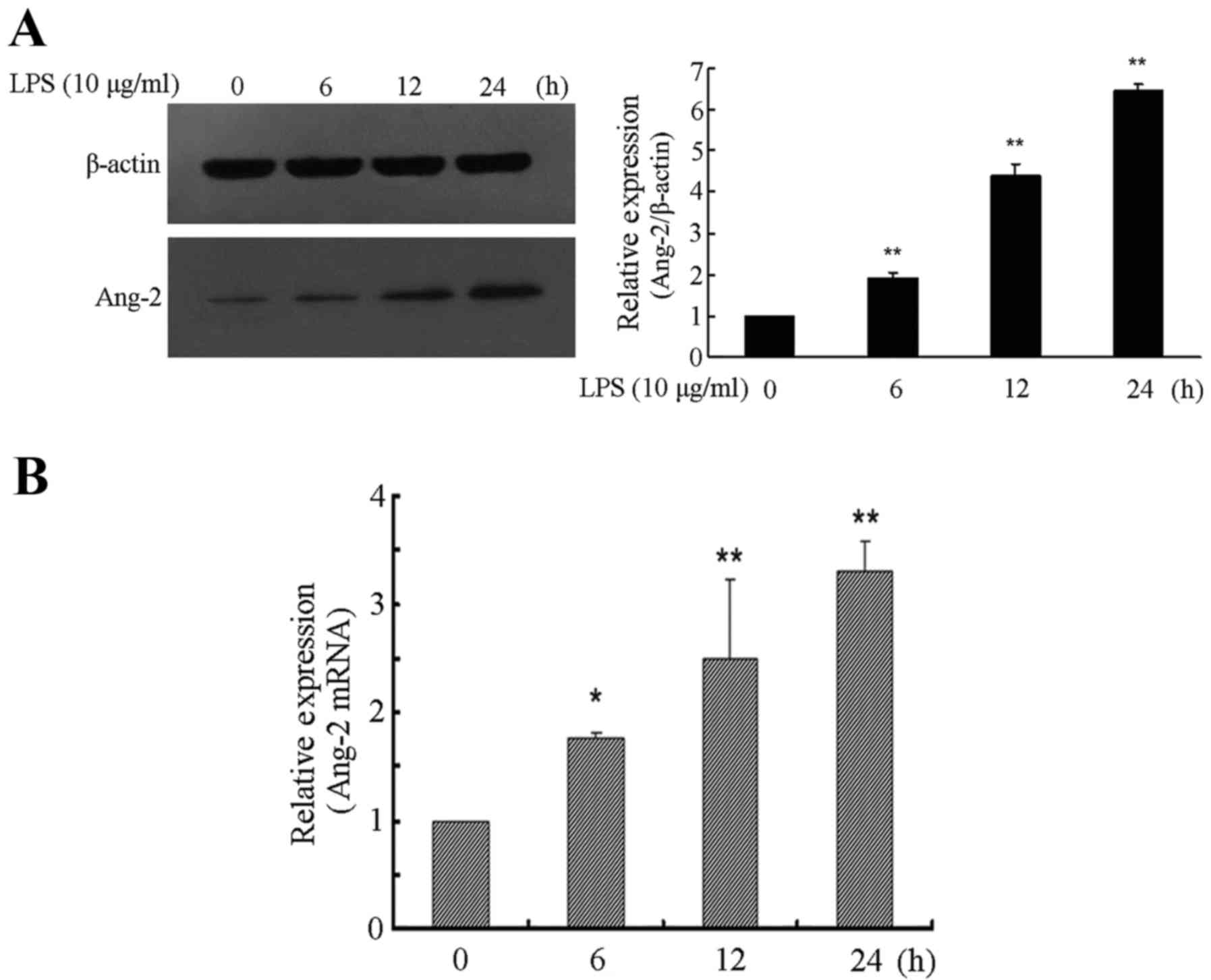
LPS time-dependently induces Ang-2
expression in RPMVECs
In the present study, RPMVECs were used to
investigate LPS-induced Ang-2 expression. The RPMVECs were cultured
in DMEM and 10% FBS medium containing 10 µg/ml LPS for 0, 6, 12 and
24 h respectively, which resulted in a time-dependent increase of
Ang-2 expression. Western blot analysis showed that expression
level of Ang-2 was significantly higher in cells incubated for 6,
12 and 24 h with LPS compared with cells incubated without LPS
(P<0.01). Following 24 h of exposure to LPS, the expression
levels of Ang-2 exhibited a >6-fold increase compared with the
control group (P<0.01; Fig. 1A).
The relative quantity of Ang-2 mRNA was also determined and was
>3-fold higher at 24 h following LPS induction compared with
that in the control cultures (P<0.01; Fig. 1B).
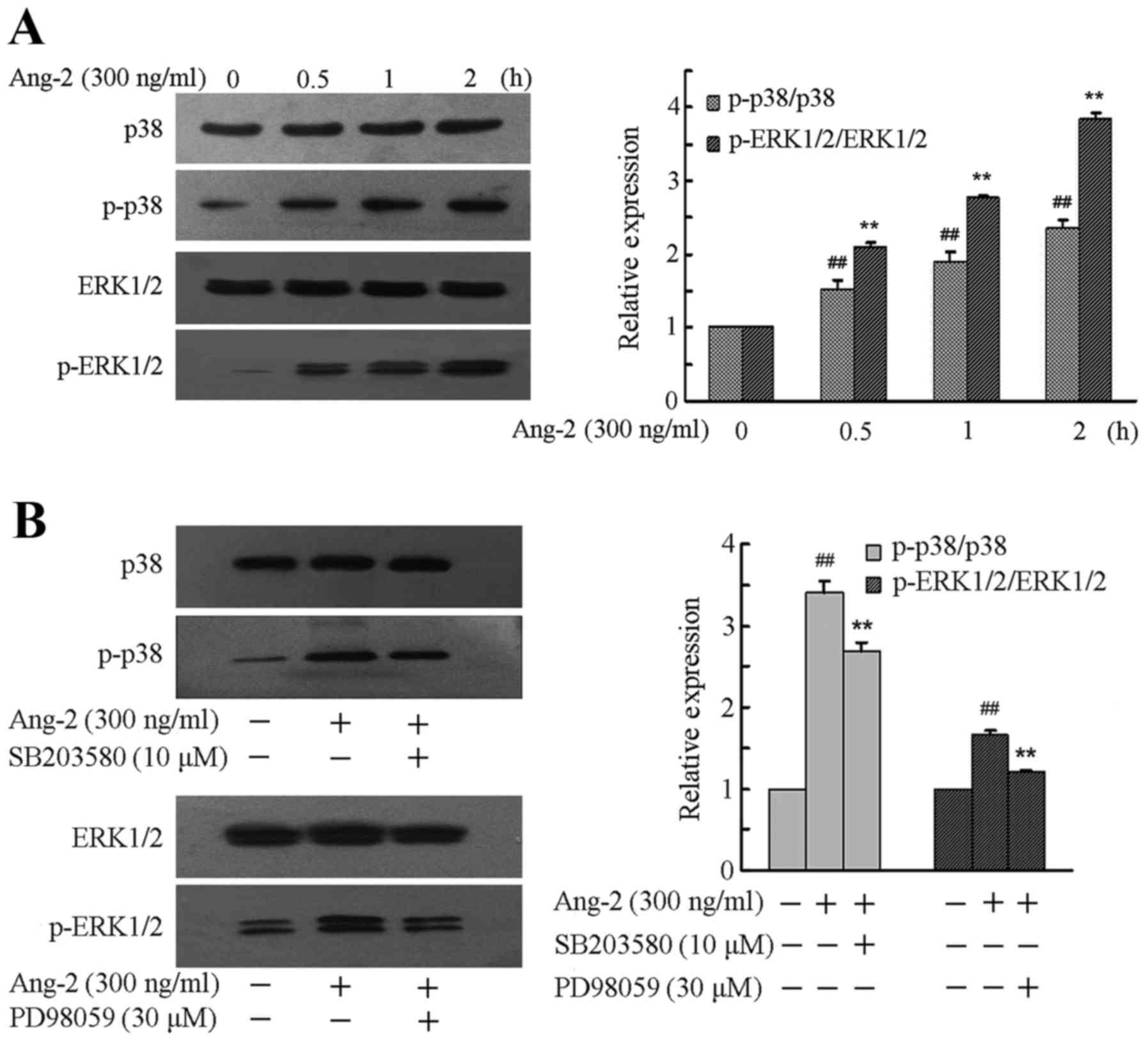
Ang-2 evokes MAPK activation and its
inhibitors modulate the expression of MAPKs
To confirm whether the MAPK pathway is involved in
the LPS-induced Ang-2-mediated apoptosis of RPMVECs, the expression
levels of p38 and ERK1/2 and their phosphorylation levels when
RPMVECs were treated with Ang-2 were first investigated, and then
selective inhibitors of MAPKs were used to further confirm the
roles of the p38 and ERK1/2 pathways. RPMVECs were incubated in
DMEM and 10% FBS medium containing 300 ng/ml Ang-2 for 0–2 h. A
significant time-dependent increase was observed in the expression
levels of p-P38 and p-ERK1/2 compared with that prior to treatment
(P<0.01; Fig. 2A). To further
clarify whether Ang-2 induced the phosphorylation of MAPKs, RPMVECs
were cultured in DMEM and 10% FBS medium and treated with SB203580
(10 µM) and PD98059 (30 µM) for 2 h followed by Ang-2 (300 ng/ml)
treatment for 1 h. Western blotting results revealed that
pre-incubation with these inhibitors significantly prevented the
phosphorylation of p38 and ERK1/2 compared with that in the RPMVECs
treated with Ang-2 alone (P<0.01; Fig. 2B).
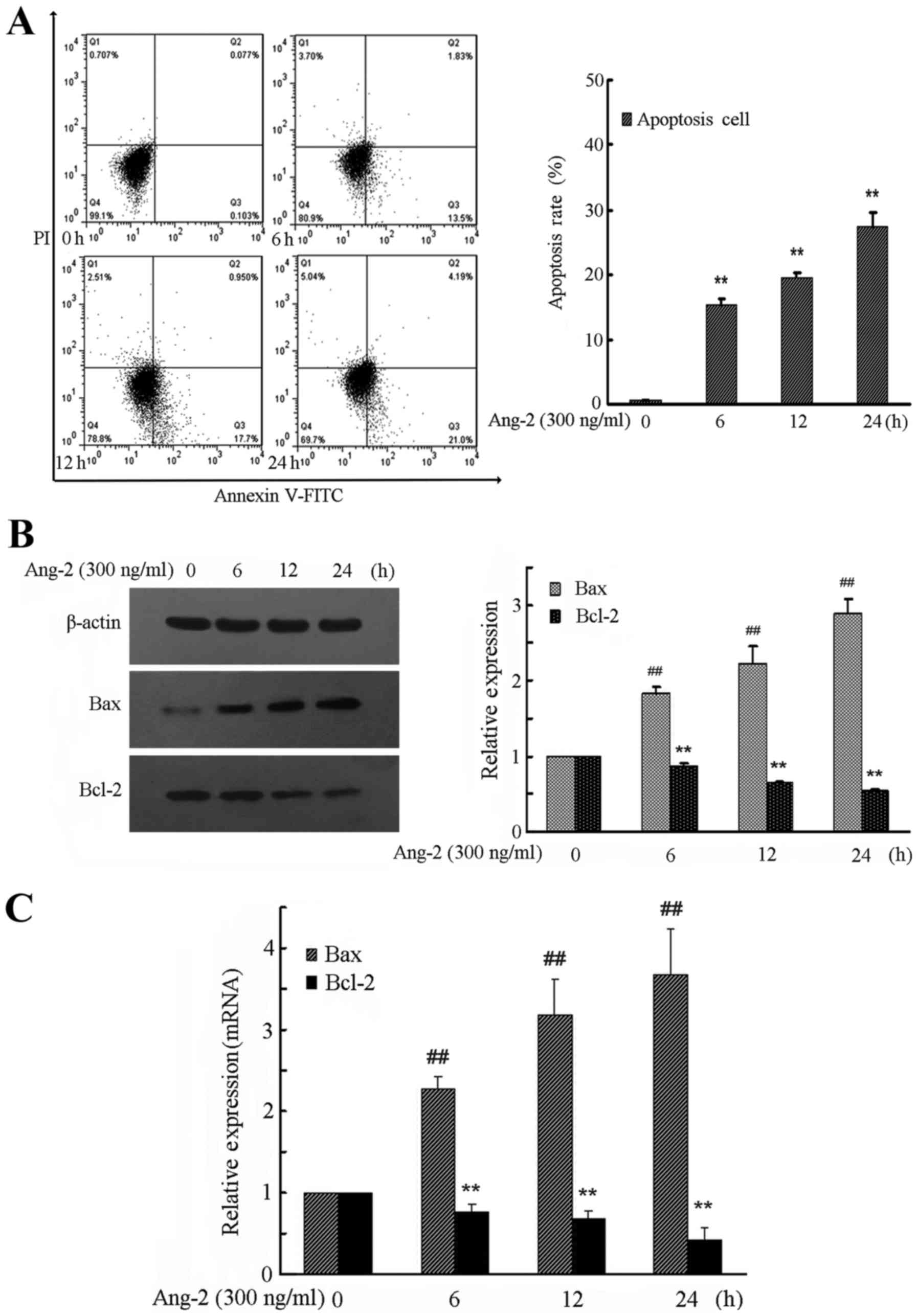
Ang-2 time-dependently mediates the
apoptosis of RPMVECs
To demonstrate how Ang-2 induces cell apoptosis,
flow cytometry with Annexin V-FITC and PI double staining was
performed. The results demonstrated a significant increase in the
numbers of apoptotic and necrotic cells in the Ang-2 group compared
with the control group (Fig. 3A).
RPMVECs treated with Ang-2 demonstrated a time-dependent increase
in apoptosis rate compared with the control group (P<0.01).
To investigate the expression levels of
apoptosis-related proteins, the downstream molecules Bax and Bcl-2
were investigated. In Ang-2-treated RPMVECs, activation of MAPK
pathways led to a time-dependent downregulation of antiapoptotic
Bcl-2 expression levels, whereas proapoptotic Bax expression levels
were time-dependently upregulated (Fig.
3B and C). The expression of Bax and Bcl-2 protein and mRNA was
significantly changed by exposure to Ang-2 compared with that in
the control group (P<0.01).
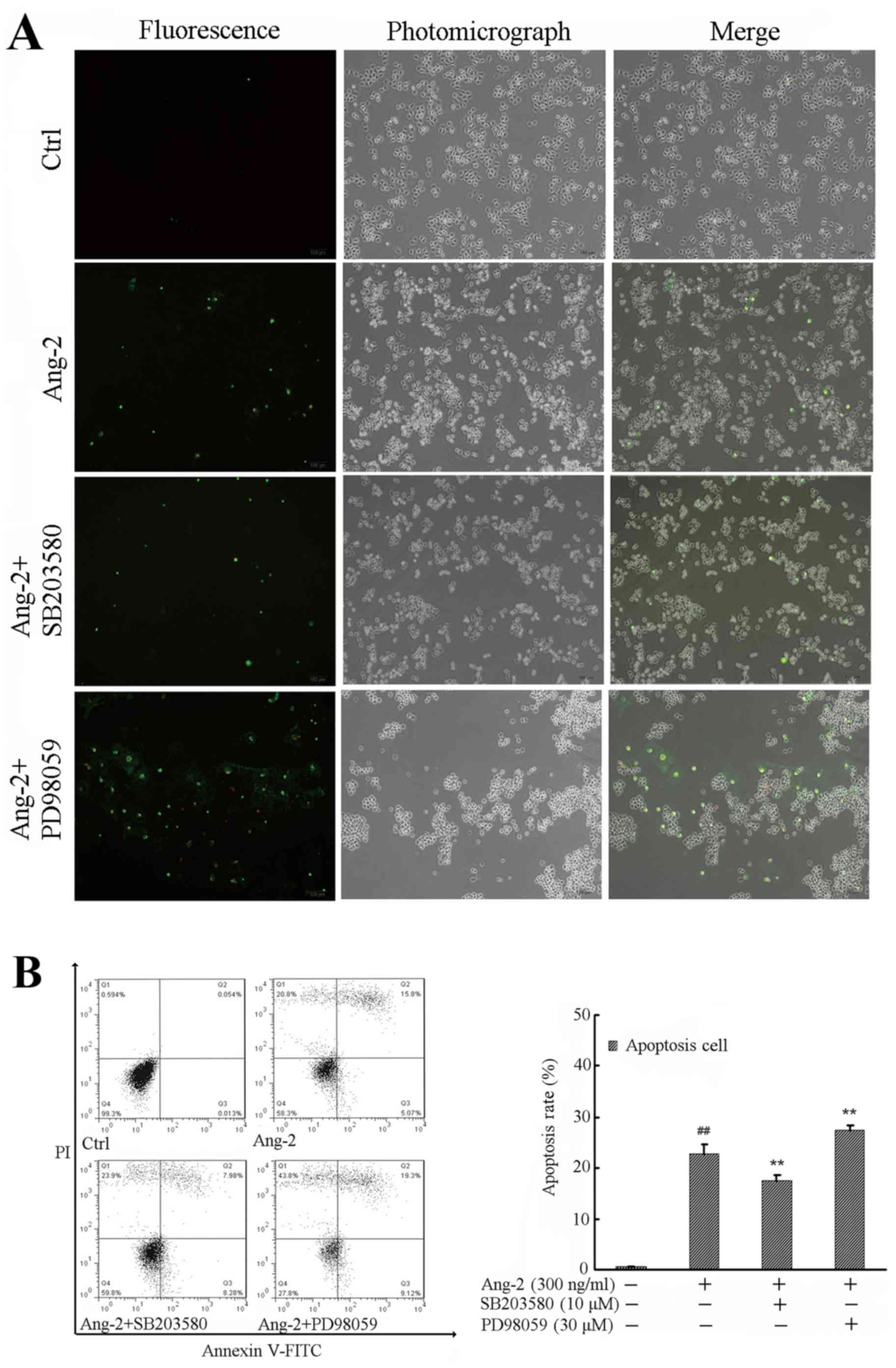
p38 and ERK1/2 signaling pathways are
involved in the Ang-2-induced apoptosis of RPMVECs
To further investigate the effects of p38 and ERK1/2
on Ang-2 induced cell apoptosis, RPMVECs were incubated with
vehicle control, Ang-2, Ang-2 + SB203580, or Ang-2 + PD98059 for 24
h. Electron microscopy revealed morphological changes following
treatment, including cells turning round and detaching from the
neighboring cells. In particular, PD98059 had an obvious effect on
cell morphology. Changes in the RPMVECs included emitting
light-green and light-red fluorescence, as observed using PI
staining and fluorescence microscopy; changes were most notable in
the PD98059-treated group (Fig. 4A).
Analysis of the cell apoptosis rate demonstrated that Ang-2
treatment increased early and late apoptotic cell rates compared
with those in the control group (P<0.01), while SB203580
pre-incubation significantly attenuated the cell apoptosis rate
compared with that in the Ang-2-alone group (P<0.01). However,
the effect of PD98059 on RPMVEC apoptosis was opposite to that of
SB203580 (Fig. 4B).
In addition, the expression levels of
apoptosis-related Bax and Bcl-2 were investigated. The expression
levels of Bax and Bcl-2 were significantly altered by Ang-2
compared with that in the control group (P<0.01); while SB203580
pretreatment attenuated Bax protein (P<0.01) and mRNA expression
levels (P<0.05), it increased the expression levels of Bcl-2
protein (P<0.01) and mRNA (P<0.05) compared with those in the
group treated with Ang-2 alone. However, PD98059 pretreatment
increased Bax protein (P<0.01) and mRNA expression (P<0.05)
and attenuated the expression levels of Bcl-2 protein (P<0.01)
and mRNA (P<0.05) compared with those in the group treated with
Ang-2 alone (Fig. 4C and D).
Discussion
The results of the current study indicate
demonstrate that Ang-2, which is induced by LPS, mediates RPMVEC
apoptosis via the MAPK signaling pathway. The main observations of
the study include: i) LPS at a concentration of 10 µg/ml
significantly promoted Ang-2 expression by endothelial cells in a
time-dependent manner. ii) Ang-2 at a concentration of 300 ng/ml
significantly increased the phosphorylation levels of p38 and
ERK1/2, elicited morphological changes in endothelial cells and
induced apoptosis-related protein expression. iii) Activation of
the p38 pathway by Ang-2 induced endothelial cell apoptosis since
the selective inhibition of this pathway by SB203580 attenuated
cell apoptosis and decreased the expression of apoptosis-related
proteins. iv) The anti-apoptotic effect of Ang-2 was mediated
through the ERK1/2 pathway because selective suppression of this
pathway by PD98059 promoted endothelial cell apoptosis and
apoptosis-related protein activation.
There is increasing evidence indicate that Ang
proteins are associated with the inflammatory response, as the
overexpression of Ang-1 has been shown to promote survival and
homodynamic functions and reduce the expression of adhesion
molecules in mice with LPS-induced ALI (16). However, studies of Ang-2 have found
that high levels disrupt the functional architecture of lung
endothelial cells, and that the barrier can be rescued with
administration of Ang-1, indicating that Ang-2 may promote
inflammation (16,17). Genetic studies have identified
polymorphisms in the ANGPT2 gene that are associated with an
increased risk of developing ALI (18). An early increase of Ang-2 levels
indicates the importance of early endothelial injury and vascular
permeability in the pathogenesis of ALI (19). Whether Ang-3 and Ang-4 regulate
inflammation has not yet been elucidated; however, Ang-3 and Ang-4
have both been observed to activate Tie-2 receptors, suggesting
that they may induce anti-inflammatory effects similar to those of
Ang-1 (20).
Despite evidence indicating the important role of
Ang in the regulation of inflammatory reaction, little is known
regarding the stimulation of endogenous Ang production by LPS. High
concentrations of LPS have been shown to stimulate numerous
endothelial responses, including the induction of apoptosis, which
may impair vascular integrity and increase the permeability of the
endothelium (21). However, the
underlying mechanism by which Ang-2 mediates LPS-induced
microvascular endothelial cell apoptosis is not fully known.
Although several studies have shown that p38 and ERK1/2 signaling
pathways are involved in the apoptosis of microvascular endothelial
cells, it is unclear whether the apoptotic effect of Ang-2 is
mediated through p38 and ERK1/2 pathways (22,23). In
the present study, the roles of p38 and ERK1/2 in the Ang-2-induced
apoptosis of RPMVECs were investigated. It was found that Ang-2
activates the phosphorylation of p38 and ERK1/2. These findings
indicate that elevated activity of p38 and ERK1/2 might be involved
in the apoptosis of endothelial cells. Therefore, inhibition of p38
MAPK and ERK1/2 pathways might ameliorate or aggravate
microvascular endothelial cell damage.
To explore the mechanisms through which Ang-2
mediates LPS-induced endothelial cell apoptosis, the role of MAPK
activation was examined by assessing the expression of the
phosphorylation of P38 and ERK1/2. Previous studies have shown that
MAPK pathways are associated with vascular inflammatory reactions
modulated by ROS (24), and
activation of MAPK proteins is vital in the cellular responses
associated with inflammatory stimuli such as LPS (25). Previous studies have demonstrated
that inactivation of ERK1/2 and activation of p38 are involved in
the induction of mitochondrial-mediated apoptosis in cancer cells
(26,27). The present study investigated whether
endothelial cells are affected similarly. The results revealed that
LPS (10 µg/ml) time-dependently increased Ang-2 protein and mRNA
expression. Furthermore, cells incubated with Ang-2 (300 ng/ml)
exhibited significantly elevated expression levels of p-p38 and
p-ERK1/2. A study by Harfouche and Hussain indicated that Ang-2 at
concentrations of 50–300 ng/ml activates Tie-2 receptors and
increases phosphorylation in the Akt, ERK1/2 and p38 MAPK pathways
while significantly inhibiting the JNK/SAPK pathway (11). In the present study, following the
selective regulation of p38 and ERK1/2 pathways, Ang-2
significantly increased Bax expression levels and suppressed Bcl-2
expression levels. Thus, it may be concluded that the
apoptosis-related proteins Bax and Bcl-2 are downstream target
proteins of p38 and ERK1/2 pathways. These results are similar to
those of other studies, which indicated that inactivation of ERK1/2
and activation of p38 could mediate the upregulation of Bax and
downregulation of Bcl-2, and finally trigger the apoptotic pathway
(28,29).
To further demonstrate the involvement of MAPK
signaling pathways in Ang-2-mediated apoptosis, inhibitors of p38
(SB203580) and ERK1/2 (PD98059) were used to explore the
association of P38 MAPK and ERK1/2 signaling pathways with the
modulation of RPMVEC apoptosis. Results indicated that the
apoptosis of RPMVECs induced by Ang-2 was significantly inhibited
by SB203580 and markedly increased by PD098059, suggesting that p38
and/or ERK1/2 may play important roles in modulating the
Ang-2-mediated apoptosis of RPMVECs. Further assessment of the
expression of apoptosis-related proteins Bax and Bcl-2 was
conducted by western blot and RT-qPCR analysis following
pre-incubation for 2 h with SB203580 (10 µM) or PD098059 (30 µM).
The p38 inhibitor significantly decreased the expression of Bax and
increased the expression of Bcl-2 mediated by Ang-2, while the
apoptotic effect of the ERK1/2 inhibitor opposes that of the p38
inhibitor. These results indicate that inhibition of the p38
pathway prevented the Ang-2-mediated apoptosis of RPMVECs, while
inhibition of the ERK1/2 pathway resulted in a pro-apoptotic
effect. This is consistent with a previous study showing that
activation of the p38 pathway by Ang-2 promotes endothelial cell
apoptosis, where selective inhibition of this signaling pathway
improved endothelial cell survival and attenuated caspase-3 and −9
activation (11). Also consistent
with this, inhibition of either p38 or ERK1/2 has been shown to
prevent TNF-α-induced increases in the permeability of human lung
microvascular endothelial cells, suggesting crucial roles for both
p38 and ERK1/2 in the microvascular endothelium (30). However, in the present study, the
reason why activation of MAPK signaling pathways by Ang-2 resulted
in cell apoptosis rather than an anti-apoptotic effect may be the
involvement of an additional cell signaling pathway, such as
PI3K/Akt, c-Jun N-terminal kinase and nuclear factor-κB, and the
cross-talk between them.
There are several limitations of the current study.
Firstly, downstream signaling from the MAPKs that may act to cause
cell apoptosis were not explored. Secondly, cross-talk among MAPKs
was not analyzed, although the activity of one MAPK can be
influenced by another or there may be interplay between NF-κB and
MAPK signaling pathways (31,32).
Thirdly, only a single RPMVEC cell line was investigated. It
remains unclear whether human pulmonary microvascular endothelial
cells would function in the same way as the RPMVECs used in the
current study.
In summary, it may be concluded that Ang-2, as the
downstream factor of LPS, could increase the LPS-induced effects on
RPMVECs. The activation of p38 MAPK and ERK1/2 plays an important
role in the Ang-2-mediated apoptosis of RPMVECs. Inhibition of the
p38 MAPK pathway exerts an anti-apoptotic effect on endothelial
cells, while inhibition of the ERK1/2 pathway exhibits a
pro-apoptotic effect. These findings imply that Ang-2 may act as an
inflammatory factor in the inflammatory injury of the microvascular
endothelium in ALI.
Acknowledgements
The authors would like to thank Professor Lin Zhang
for his guidance.
Funding
The present study was supported by the Anhui Natural
Science Foundation (grant no. 1208085QH142).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SL was involved in data collection and analysis and
contributed to writing the manuscript. MZ designed and performed
the experiments. YY performed experiments and provided guidance. LZ
contributed to experimental design and data collection.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing
interests.
References
|
1
|
Matthay MA, Ware LB and Zimmerman GA: The
acute respiratory distress syndrome. J Clin Invest. 122:2731–2740.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu H, Liang X, Wang D, Zhang H, Liu L,
Chen H, Li Y, Duan Q and Xie K: Combination therapy with nitric
oxide and molecular hydrogen in a murine model of acute lung
injury. Shock. 43:504–511. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chopra M, Reuben JS and Sharma AC: Acute
lung injury: Apoptosis and signaling mechanisms. Exp Biol Med
(Maywood). 234:361–371. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin WC, Chen CW, Huang YW, Chao L, Chao J,
Lin YS and Lin CF: Kallistatin protects against sepsis-related
acute lung injury via inhibiting inflammation and apoptosis. Sci
Rep. 5:124632015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li X, Shu R, Filippatos G and Uhal BD:
Apoptosis in lung injury and remodeling. J Appl Physiol (1985).
97:1535–1542. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stefancec T: Endothelial apoptosis: Could
it have a role in the pathogenesis and treatment of disease. Chest.
117:841–854. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsuda N, Takano Y, Kageyama S,
Hatakeyama N, Shakunaga K, Kitajima I, Yamazaki M and Hattori Y:
Sliencing of caspase-8 and caspase-3 by RNA interference prevents
vascular endothelial cell injury in mice with endotoxic shock.
Cardiovasc Res. 76:132–140. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang H, Bhat A, Woodnutt G and Lappe R:
Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer.
10:575–589. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuan HT, Khankin EV, Karumanchi SA and
Parikh SM: Angiopoietin 2 is a partial agonist/antagonist of Tie2
signaling in the endothelium. Mol Cell Biol. 29:2011–2022. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davis JS, Yeo TW, Piera KA, Woodberry T,
Celermajer DS, Stephens DP and Anstey NM: Angiopoietin-2 is
increased in sepsis and inversely associated with nitric
oxide-dependent microvascular reactivity. Crit Care. 14:R892010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harfouche R and Hussain SN: Signaling and
regulation of endothelial cell survival by angiopoietin-2. Am J
Physiol Heart Circ Physiol. 291:H1635–H1645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kumpers P, van Meurs M, Molema G, Molema
G, Bijzet J, Lukasz A, Biertz F, Haller H and Zijlstra JG: Time
course of angiopoietin-2 release during experimental human
endotoxemia and sepsis. Critical Care. 13:R642009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harfouche R, Gratton JP, Yancopoulous GD,
Noseda M, Karsan A and Hussain SN: Angiopoietin-1 activates both
anti- and proapoptotic mitogen-activated protein kinases. FASEB J.
17:1523–1525. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harfouche R, Hasséssian HM, Guo Y, Faivre
V, Srikant CB, Yancopoulos GD and Hussain SN: Mechanisms which
mediate the antiapoptotic effects of angiopoietin-1 on endothelial
cells. Microvasc Res. 64:135–147. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thurston G, Wang Q, Baffert F, Rudge J,
Papadopoulos N, Jean-Guillaume D, Wiegand S, Yancopoulos GD and
McDonald DM: Angiopoietin 1 causes vessel enlargement, without
angiogenic sprouting, during a critical developmental period.
Development. 132:3317–3326. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gallagher DC, Parikh SM, Balonov K, Miller
A, Gautam S, Talmor D and Sukhatme VP: Circulating angiopoietin 2
correlates with mortality in a surgical population with acute lung
injury/adult respiratory distress syndrome. Shock. 29:656–661.
2008.PubMed/NCBI
|
|
18
|
Meyer NJ, Li M, Feng R, Bradfield J,
Gallop R, Bellamy S, Fuchs BD, Lanken PN, Albelda SM, Rushefski M,
et al: ANGPT2 genetic variant is associated with trauma-associated
acute lung injury and altered plasma angiopoietin-2 isoform ratio.
Am J Respir Crit Care Med. 183:1344–1353. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Agrawal A, Matthay MA, Kangelaris KN,
Stein J, Chu JC, Imp BM, Cortez A, Abbott J, Liu KD and Calfee CS:
Plasma angiopoietin-2 predicts the onset of acute lung injury in
critically Ill patients. Am J Respir Crit Care Med. 187:736–742.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee HJ, Cho CH, Hwang SJ, Choi HH, Kim KT,
Ahn SY, Kim JH, Oh JL, Lee GM and Koh GY: Biological
characterization of angiopoietin-3 and angiopoietin-4. FASEB J.
18:1200–1208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mizumura K, Gon Y, Kumasawa F, Onose A,
Maruoka S, Matsumoto K, Hayashi S, Kobayashi T and Hashimoto S:
Apoptosis signal-regulating kinase 1-mediated signaling pathway
regulates lipopolysaccharide-induced tissue factor expression in
pulmonary microvasculature. Int Immunopharmacol. 10:1062–1067.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tan J, Liu D, Lv X, Wang L, Zhao C, Che Y,
Xie Q and Cui X: MAPK mediates inflammatory response and cell death
in rat pulmonary microvascular endothelial cells in an
ischemia-reperfusion model of lung transplantation. J Heart Lung
Transplant. 32:823–831. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Z, Zhang J, Li B, Mao W and Chen S:
MAPK signaling mediates low shear stress-induced oxidative damage
in human umbilical vein endothelia cells in vitro. Nan Fang Yi Ke
Da Xue Xue Bao. 34:603–608. 2014.PubMed/NCBI
|
|
24
|
Rashed LA, Hashem RM and Soliman HM:
Oxytocin inhibits NADPH oxidase and P38 MAPK in cisplatin-induced
nephrotoxicity. Biomed Pharmacother. 65:474–480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chi G, Wei M, Xie X, Soromou LW, Liu F and
Zhao S: Suppression of MAPK and NF-κB pathways by limonene
contributes to attenuation of lipopolysaccharide-induced
inflammatory responses in acute lung injury. Inflammation.
36:501–511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Z, Miao L, Lv C, Sun H, Wei S, Wang
B, Huang C and Jiao B: Wentilactone B induces G2/M phase arrest and
apoptosis via the Ras/Raf/MAPK signaling pathway in human hepatoma
SMMC-7721 cells. Cell Death Dis. 4:e6572013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Min L, He B and Hui L: Mitogen-activated
protein kinases in hepatocellular carcinoma development. Semin
Cancer Biol. 21:10–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ye Y, Hou R, Chen J, Mo L, Zhang J, Huang
Y and Mo Z: Formononetin-induced apoptosis of human prostate cancer
cells through ERK1/2 mitogen-activated protein kinase inactivation.
Horm Metab Res. 44:263–267. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nwariaku FE, Rothenbach P, Liu Z, Zhu X,
Turnage RH and Terada LS: Rho inhibition decreases TNF-induced
endothelial MAPK activation and monolayer permeability. J Appl
Physiol (1985). 95:1889–1895. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hoefen RJ and Berk BC: The role of MAP
kinases in endothelial activation. Vascul Pharmacol. 38:271–273.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kanaji N, Sato T, Nelson A, Wang X, Li Y,
Kim M, Nakanishi M, Basma H, Michalski J, Farid M, et al:
Inflammatory cytokines regulate endothelial cell survival and
tissue repair functions via NF-κB signaling. J Inflamm Res.
4:127–138. 2011.PubMed/NCBI
|