Introduction
Diabetic nephropathy (DN) is one of the main causes
of end-stage renal disease worldwide (1) Typical morphlogical changes of DN
include proliferation of mesangial cells (MCs) and accumulation of
the extracellular matrix (ECM), which contributes to the thickening
of basement membranes and glomerulosclerosis (2,3). It is
therefore important to develop effective therapeutic approaches for
the treatment and prevention of glomerulosclerosis in diabetes,
which may include inhibition of MC proliferation and ECM
accumulation.
Hyperglycemia and inflammation serve roles in the
initiation of DN (4). Inflammation
and pro-inflammatory cytokines are critical to the pathogenesis of
DN (5). Nuclear factor κB (NF-κB) is
considered to be a major signaling pathway associated with
inflammation, regulating several inflammatory response genes
including monocyte chemoattractant protein-1 (MCP-1) and
transforming growth factor (TGF)β1 (5). MCP-1 and TGF-β1 are secreted by
glomerular MCs and are inflammatory mediators in DN (5,6). MCP-1,
TGF-β1 and fibronectin (FN) secretion can be enhanced via NF-κB
activation, leading to MC proliferation, abnormal ECM accumulation
and tubulointerstitial sclerosis (7,8).
Adenosine monophosphate-activated protein kinase
(AMPK) is an enzyme associated with cellular energy homeostasis
(9). Exposure to high glucose (HG)
concentrations suppresses AMPK activation (10–14),
inhibiting the activation of NF-κB (15). In addition, inactivation of AMPK is
associated with pro-inflammatory and pro-fibrotic damage (16).
Thalidomide (Thd) was previously withdrawn from the
market due to the high incidence of teratogenicity (17). However, the return of Thd in clinical
practice is primarily due to its anti-inflammatory, anti-fibrotic
and immune-modulatory properties, as it is currently used for the
treatment of inflammatory bowel disease, multiple myeloma and
rheumatic disease (17). Thd can
suppress the levels of pro-inflammatory cytokines, which include
tumor necrosis factor α, interleukin (IL)-1β, IL-6 and TGF-β1
(17–19). Previous studies have demonstrated the
anti-inflammatory effects of Thd in diabetic retinopathy,
neuropathy and cardiomyopathy animal models (20–22). A
previous study demonstrated the renal-protective effect of Thd in
DN rats through the activation of AMPK and inhibition of
NF-κB/MCP-1 and TGF-β1/mothers against decapentaplegic homolog
(Smad) signaling pathways (23). The
association between Thd-induced AMPK activation and the inhibition
of HG-induced MC proliferation and ECM accumulation via NF-κB and
TGF-β1 signaling remains unknown. The aim of the present study was
to demonstrate the effects of Thd on cell proliferation and ECM
expression in HG-cultured MCs and the underlying mechanisms.
Materials and methods
Cell culture
Human mesangial cell line T-SV40 was donated by Dr
Li Xuewang at Union Medical College Hospital (Beijing, China).
Cells were cultured in Dulbecco's modified Eagle's media (DMEM;
HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10% fetal bovine serum (FBS; HyClone; GE Healthcare Life
Sciences) and maintained at 37°C in a 5% CO2-humidified
incubator. Medium was changed every 2 days and only cells at
passage 3–5 were subsequently used. Cells were synchronized prior
to use in subsequent experiments following pre-incubation with
Minimum Essential Medium (MEM; HyClone; GE Healthcare Life
Sciences) supplemented with 1% FBS at 37°C overnight. Cells were
subsequently exposed to Normal glucose (NG) containing 5.6 mmol/l
D-glucose and HG containing 30 mmol/l D-glucose with additional
administration of Thd (0, 10, 50, 100 and 200 µg/ml) at 37°C for
12, 24 or 48 h.
Reagents
Thd (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
was dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich) and
diluted in saline to a final concentration of 100 mg/ml stock
solution. In the Thd-treated groups, the Thd stock solution was
diluted in DMSO to a final concentration of 0.2%. The control
groups received the corresponding volume of vehicle control (DMSO
only). AMPK inhibitor (compound C) was purchased from Merck KGaA
and AMPK agonist (5-aminoimidazole-4-carboxamide
1β-D-ribofuranoside; AICAR) was purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). For compound C (C) or AICAR
(A) treatment, synchronized MCs were treated with or without 10 µM
C or 1 mM A under NG at 37°C for 1 h. Following inhibitor or
activator incubation, cells were treated under HG with or without
Thd (100 µg/ml) at 37°C for 24 h.
MTT assay
Cell proliferation was measured by MTT assay.
Following 12, 24 or 48-h treatment with various concentrations (0,
10, 50, 100 and 200 µg/ml) of Thd, MCs were seeded into 96-well
plates at a density of 5×103 cells/well. Following
incubation, 20 µl MTT (5 mg/ml; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was added to each well and
further incubated at 37°C for 4 h. Following incubation, the medium
was removed and 150 µl DMSO was added to each sample. Cell
proliferation was determined by measuring the absorbance at 560 nm
using a microplate reader (Model 550; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The mean optical density was determined by
examining seven wells per group.
Bromodeoxyuridine (BrdU) assay
The quantification of MC proliferation was based on
the measurement of BrdU incorporation during DNA synthesis.
Following 12, 24 or 48-h treatment with various concentrations (0,
10, 50, 100 and 200 µg/ml) of Thd, MCs were seeded into 96-well
plates, as described above. The cells were subsequently treated
with BrdU during the final 2 h of incubation. The BrdU Cell
Proliferation assay kit (cat. no. 2750; EMD Millipore, Billerica,
MA, USA) was performed according to the manufacturer's
protocol.
ELISA
Cell supernatant was harvested from the NG group and
different treatment groups under HG with Thd (0, 10, 50, 100 and
200 µg/ml) and centrifuged at 1,500 × g for 10 min at 4°C. The
level of TGF-β1 (cat. no. EK0513), MCP-1 (cat. no. EK0441) and FN
(cat. no. EK0349) secreted were detected using ELISA kits (all
Boster Biological Engineering Co., Wuhan, China) (24). Absorbance was measured at 450 nm
using a microplate reader (Model 550; Bio-Rad Laboratories, Inc.).
Each sample was analyzed in triplicate.
Western blot analysis
MCs were washed with PBS. Total protein was
extracted from MCs using radioimmunoprecipitation assay buffer
(Beyotime Institute of Biotechnology, Haimen, China) supplemented
with protease (cat. no. HY-K0011) and phosphatase inhibitors (cat.
no. HY-K0021; each MedChemExpress, Monmouth Junction, NJ, USA),
which were added prior to use. Total protein was quantified using a
bicinchoninic acid assay (Beyotime Institute of Biotechnology) and
50 µg protein/lane was separated via SDS-PAGE on a 12% gel. The
separated proteins were subsequently transferred onto
nitrocellulose membranes (Whatman International Ltd., Maidstone,
Kent, UK) and blocked using blocking buffer (PBS and 0.1% Tween-20)
with 5% non-fat milk for 1 h at room temperature. Then membranes
were incubated with primary antibodies against TGF-β1 (1:1,000;
cat. no. ab92486), MCP-1 (1:1,000; cat. no. ab9669), inhibitor of
NF-κB (IκBα) (1:1,000; cat. no. ab32518), β-actin (1:1,000; cat.
no. ab8226; all Abcam, Cambridge, UK), NF-κB (1:1,000; cat. no.
4764), AMPK (1:1,000; cat. no. 2532), phosphorylated AMPK (p-AMPK;
1:1,000; cat. no. 50081), tubulin (1:1,000; cat. no. 2148) and
GAPDH (1:1,000; cat. no. 5174; all Cell Signaling Technology, Inc.)
overnight at 4°C. Membranes were washed three times with
Tris-buffered saline containing Tween®−20. Membranes
were incubated with corresponding horseradish peroxidase-conjugated
anti-rabbit immunoglobulin G (IgG; 1:5,000; cat. no. sc2370) or
anti-mouse IgG (1:2,000; cat. no. sc2380) secondary antibodies
(each, Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 90 min
at room temperature. The membranes were then reacted with an
ECL-plus chemiluminescent detection HRP reagent (Beyotime Institute
of Biotechnology). Protein expression was quantified using the
Quantity One analysis system (version 4.62; Bio-Rad Laboratories,
Inc.).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. All statistical analyses were performed using SPSS
software (version 19.0; IBM Corp., Armonk, NY, USA). Differences
between groups were measured using a one-way analysis of variance
followed by a post hoc Bonferroni correction test. P<0.05 was
considered to indicate a statistically significant difference.
Results
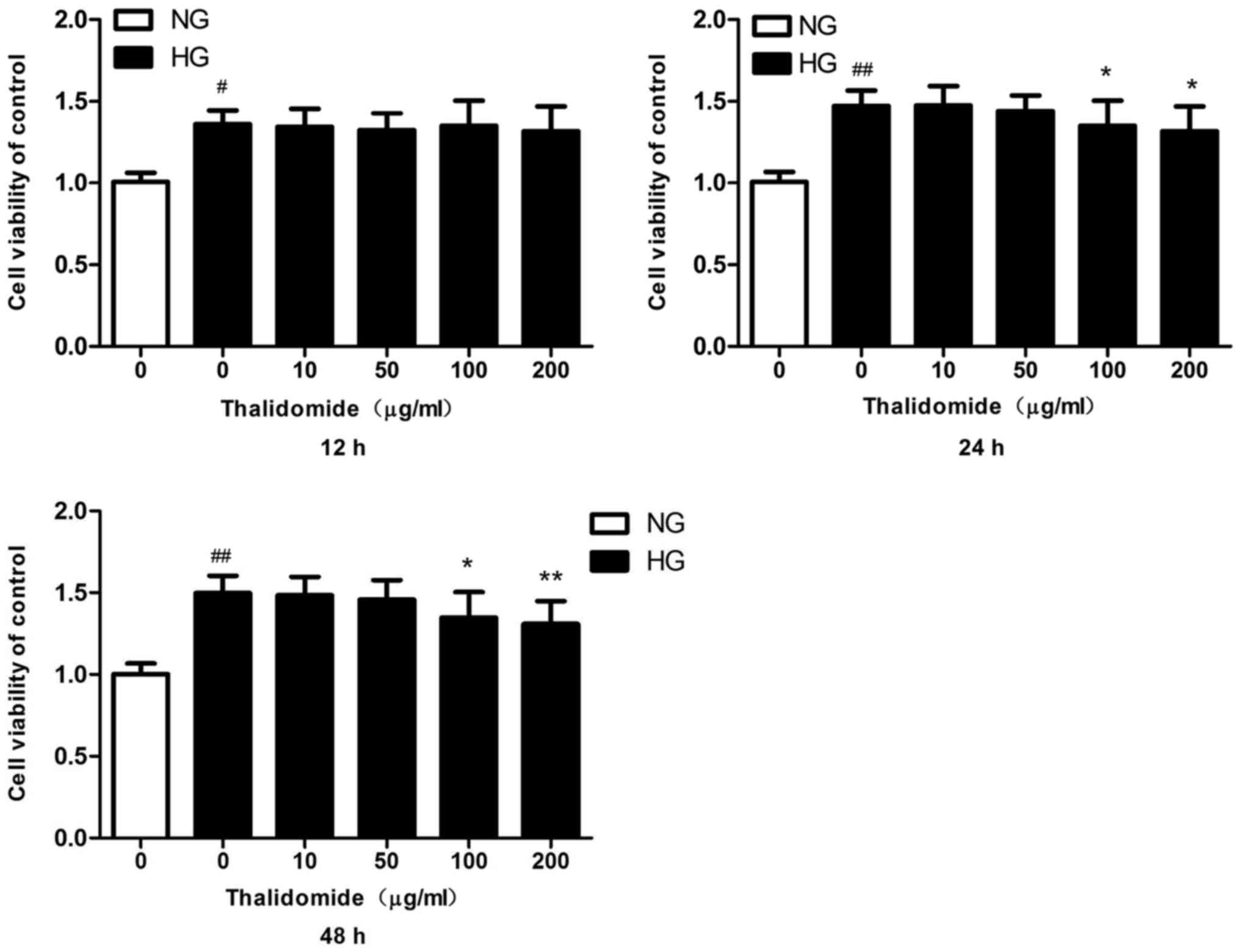
In vitro dosage of Thd in human
MCs
To determine the in vitro dosage of Thd in
MCs, cell viability was measured by MTT assay. The MTT assay was
performed following treatment with various concentrations (0, 10,
50, 100 and 200 µg/ml) of Thd for 12, 24 or 48 h. Cell viability
significantly increased in HG-cultured MCs compared with
NG-cultured MCs for 12, 24 and 48 h (Fig. 1). Cell viability significantly
decreased in HG-cultured MCs following treatment with Thd 100 and
200 µg/ml for 24 h compared with the HG-cultured MC group. In
addition, similar results were observed in HG-cultured MCs
following treatment with Thd for 48 h. There was no significant
cytotoxic effect observed in HG-induced MCs following treatment
with Thd for 12 h.
BrdU assays were performed to examine the effects of
Thd on MC cell proliferation. BrdU incorporation significantly
increased in HG-cultured MCs compared with NG-cultured MCs for 12,
24 and 48 h (Fig. 2). In addition,
BrdU incorporation significantly decreased in HG-cultured MCs
following treatment with Thd 100 µg/ml for 24 h compared with the
HG-cultured MC group. Therefore, treatment with Thd 100 µg/ml for
24 h in HG-cultured MCs was used in all subsequent experiments.
 | Figure 2.Effects of Thd on HG-induced MC
proliferation by BrdU assay. NG-cultured MCs were grown in DMEM
containing 5.6 mmol/l D-glucose, whilst HG-cultured MCs were grown
in DMEM containing 30 mmol/l D-glucose and following treatment with
various concentrations (0, 10, 50, 100 and 200 µg/ml) of Thd for
12, 24 or 48 h the BrdU assay was used to quantify cell
proliferation. Data are presented as the mean ± standard error of
the mean (n=5). #P<0.01 and ##P<0.001
vs. NG group; *P<0.01 and **P<0.001 vs. HG group. Thd,
thalidomide; HG, high glucose; MC, mesangial cell; BrdU,
bromodeoxyuridine; DMEM, Dulbecco's modified Eagle's medium; NG,
normal glucose. |
Effect of Thd on TGF-β1, FN and MCP-1
expression
The expression levels of TGF-β1, FN and MCP-1 were
analyzed in MCs by ELISA. The protein expression levels of TGF-β1,
FN and MCP-1 were significantly enhanced in HG-cultured MCs
compared with NG-cultured MCs (Fig.
3). However, the expression levels significantly decreased in
HG-cultured MCs following treatment with high concentrations of Thd
(100 and 200 µg/ml) compared with the HG-cultured MC group.
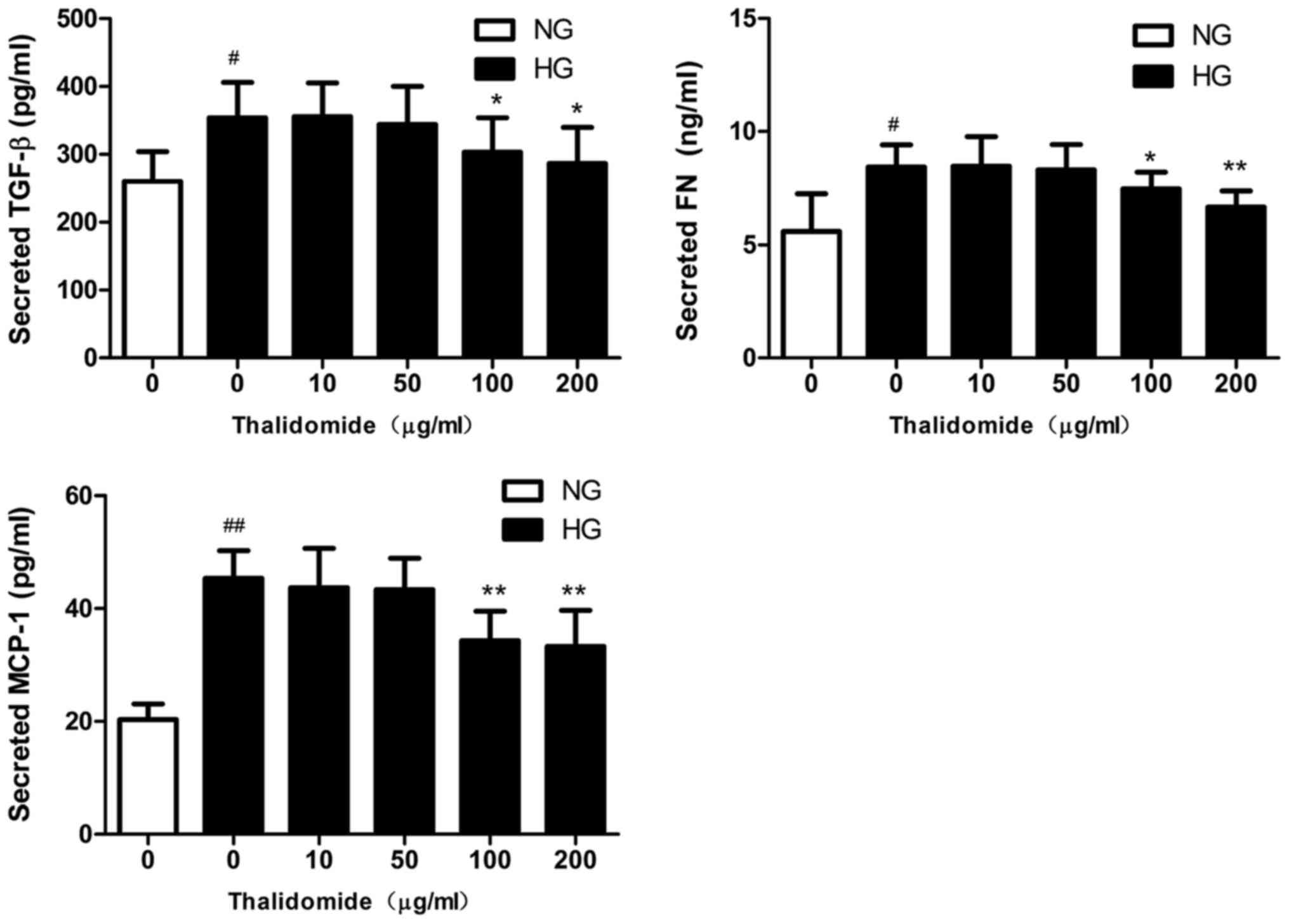 | Figure 3.TGF-β1, FN and MCP-1 protein
expression in MC supernatant. Following treatment with various
concentrations (0, 10, 50, 100 and 200 µg/ml) of Thd for 24 h, the
protein expression levels of TGF-β1, FN and MCP-1 were analyzed by
ELISA. Data are presented as the mean ± standard error of the mean
(n=3). #P<0.01 and ##P<0.001 vs. NG
group; *P<0.01 and **P<0.001 vs. HG group. TGF-β1,
transforming growth factor β1; FN, fibronectin; MCP-1, monocyte
chemoattractant protein-1; MC, mesangial cell; Thd, thalidomide;
NG, normal glucose; HG, high glucose. |
Thd modulates the expression of NF-κB,
IκBα, MCP-1 and TGF-β1 in HG-induced MCs
To investigate the potential involvement of the
NF-κB/MCP-1 signaling pathways in the regulation of HG-induced cell
proliferation, the protein expression levels of NF-κB, IκBα, MCP-1
and TGF-β1 were determined by western blot analysis (Fig. 4A). The expression levels of
inflammatory cytokines NF-κB, MCP-1 and TGF-β1 were significantly
increased whereas the expression level of IκBα was significantly
decreased in HG-cultured MCs compared with NG-cultured MCs
(Fig. 4B). In addition, treatment
with Thd significantly decreased the expression levels of
HG-induced inflammatory cytokines NF-κB, MCP-1 and TGF-β1 whereas
the expression level of IκBα was significantly increased in
HG-cultured MCs (Fig. 4B). These
results suggest that Thd may exert anti-inflammatory effects on
HG-induced MCs.
 | Figure 4.Effects of Thd on NF-κB, IκBα, MCP-1
and TGF-β1 expression. (A) The protein expression levels of NF-κB,
IκBα, MCP-1 and TGF-β1 were determined using western blot analysis
in NG-cultured and HG-cultured MCs following treatment with Thd
(100 or 200 µg/ml) for 24 h. (B) Quantification of NF-κB, IκBα,
MCP-1 and TGF-β1 protein expression. Data are presented as the mean
± standard error of the mean (n=4). #P<0.01 vs. NG
group and *P<0.01 vs. HG group. Thd, thalidomide; NF-κB, nuclear
factor κB; IκBα, inhibitor of NF-κB; MCP-1, monocyte
chemoattractant protein-1; TGF-β1, transforming growth factor β1;
MC, mesangial cell; NG, normal glucose; HG, high glucose. |
Thd attenuates HG-induced NF-κB and
TGF-β1 through AMPK signaling
The protein expression levels of AMPK and p-AMPK
were determined by western blot analysis in MCs (Fig. 5A). The expression level of p-AMPK was
significantly decreased in HG-cultured MCs compared with
NG-cultured MCs (Fig. 5B). In
addition, treatment with high concentrations of Thd (100 and 200
µg/ml) significantly increased the expression level of p-AMPK
(Thr172) compared with the HG-cultured MC group (Fig. 5B). Furthermore, treatment with Thd
appeared to activate AMPK and inhibit TGF-β1 and NF-κB protein
expression in a similar manner to AICAR. By contrast, compound C
appeared to reverse Thd-induced AMPK activation and enhance TGF-β1
and NF-κB protein expression (Figs.
6 and 7).
Effects of the AMPK signaling pathway
on cell viability and the cytotoxic potential of Thd
In order to examine the effects of the AMPK
signaling pathway in HG-cultured MCs, cell proliferation was
measured by MTT assay. Cell viability was significantly decreased
in HG-cultured MCs following treatment with Thd 100 µg/ml compared
with the HG-cultured MC group. Similarly, cell viability
significantly decreased in HG-cultured MCs following treatment with
AICAR for 12 or 24 h compared with the HG-cultured MC group.
However, the Thd-induced decrease in cell viability was
significantly reversed in HG-cultured MCs following treatment with
compound C (Fig. 8A). In addition,
there was no significant cytotoxic effect observed in NG-induced
MCs following treatment with Thd (100 µg/ml) for 12, 24 or 48 h
(Fig. 8B).
Discussion
The current study is the first to identify the
protective effects of Thd against HG-induced matrix protein
synthesis in human mesangial cells, to the best of our knowledge.
Following 24-h Thd incubation, HG-induced inflammatory cytokines
and abnormal ECM protein accumulation were significantly decreased
compared with the HG group. These protective effects may be due to
the upregulation of p-AMPK and inhibition of the NF-κB/MCP-1
signaling pathways.
Chronic inflammation serves a role in the initiation
of DN (4,5). Several inflammatory cytokines including
adhesion molecules and pro-inflammatory cytokines, are thought to
be involved in the pathogenesis of DN (4,25).
NF-κB, a major signaling pathway in inflammation, is activated in
response to cellular stress which includes hyperglycemia, obesity
and oxidative stress (26). NF-κB
activation induces the transcription of several target genes
including MCP-1, TNF-α and the interleukin system (26). MCP-1 can accelerate the recruitment
of macrophages to the kidneys following renal injury in DN, whereas
TGF-β1 upregulation can inhibit the accumulation of ECM in DN
(27–29). In the current study, the expression
levels of the inflammatory cytokines NF-κB, MCP-1 and TGF-β1
significantly increased in HG-induced MCs, and significantly
decreased following treatment with Thd. By contrast, treatment with
Thd significantly increased the expression level of IκBα. These
results suggest that Thd may exert anti-inflammatory effects
through inhibition of the NF-κB signaling pathway.
Previous studies have demonstrated that AMPK
activation in the diabetic kidney can be inhibited via multiple
mechanisms (30,10,11).
Studies have revealed that the downregulation of p-AMPK aggravates
the abnormal accumulation of ECM proteins and glomerular
hypertrophy via activation of the mammalian target of rapamycin and
the TGF-β1/Smad4 signaling pathway (30,31).
AMPK activation serves a role in the downregulation of TGF-β1 in DN
(32). Previous studies have
demonstrated that AMPK activation can inhibit the activation of
NF-κB (12,13). AMPK could therefore be used as a
target for the treatment of DN. In the present study, treatment
with high concentrations of Thd (100 and 200 µg/ml) for 24 h
significantly increased the expression level of p-AMPK (Thr172) and
significantly decreased the expression levels of inflammatory
cytokines NF-κB, MCP-1 and TGFβ compared with the HG-cultured MC
group. Furthermore, treatment with Thd appeared to activate AMPK
and inhibit TGF-β1 and NF-κB protein expression in a similar manner
to AICAR. Incubation with compound C appeared to reverse
Thd-induced AMPK activation and enhance NF-κB, MCP-1 and TGF-β1
protein expression. Taken together, these results suggest that the
anti-inflammatory effects of Thd depend on AMPK signaling. However,
further studies are required to demonstrate the mechanisms of Thd
in the AMPK signaling pathway.
In conclusion, the current study is the first in
vitro study to demonstrate that Thd reduces inflammation via
the activation of AMPK, and the inhibition of the NF-κB/MCP-1
signaling pathway in HG-induced MCs, to the best of our knowledge.
These results suggest that Thd may have therapeutic potential in
diabetic renal injury. However, further studies are required to
elucidate the molecular mechanism underlying Thd-induced AMPK
activation in DN.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Basic
Research Project of Shanxi Province (grant no. 201701D111001).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HXZ and RSL designed the study and provided research
funding. JY and YFL designed the experiments and provided technical
guidance. HXZ and JY performed the experiments. HXZ and JY wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
International Diabetes Federation: IDF
Diabetes Atlas 7th edition (2015). https://www.idf.org/e-library/epidemiology-research/diabetes-atlas/13-diabetes-atlas-seventh-edition.htmlAugust
14–2018
|
|
2
|
Kolset SO, Reinholt FP and Jenssen T:
Diabetic nephropathy and extracellular matrix. J Histochem
Cytochem. 60:976–986. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mason RM and Wahab NA: Extracellular
matrix metabolism in diabetic nephropathy. J Am Soc Nephrol.
14:1358–1373. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wada J and Makino H: Inflammation and the
pathogenesis of diabetic nephropathy. Clin Sci (Lond). 124:139–152.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Navarro-González JF, Mora-Fernández C, de
Fuentes Morus M and García-Pérez J: Inflammatory molecules and
pathways in the pathogenesis of diabetic nephropathy. Nat Rev
Nephrol. 7:327–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Min D, Lyons JG, Bonner J, Twigg SM, Yue
DK and McLennan SV: Mesangial cell-derived factors alter monocyte
activation and function through inflammatory pathways: Possible
pathogenic role in diabetic nephropathy. Am J Physiol Renal
Physiol. 297:F1229–F1237. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ingaramo PI, Ronco MT, Francés DE, Monti
JA, Pisani GB, Ceballos MP, Galleano M, Carrillo MC and Carnovale
CE: Tumor necrosis factor alpha pathways develops liver apoptosis
in type 1 diabetes Mellitus. Mol Immunol. 48:1397–1407. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Giunti S, Tesch GH, Pinach S, Burt DJ,
Cooper ME, Cavallo-Perin P, Camussi G and Gruden G: Monocyte
chemoattractant protein-1 has prosclerotic effects both in a mouse
model of experimental diabetes and in vitro in human mesangial
cells. Diabetologia. 51:198–207. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kemp BE, Stapleton D, Campbell DJ, Chen
ZP, Murthy S, Walter M, Gupta A, Adams JJ, Katsis F, van Denderen
B, et al: AMP-activated protein kinase, super metabolic regulator.
Biochem Soc Trans. 31:162–168. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Decleves AE, Mathew AV, Cunard R and
Sharma K: AMPK mediates the initiation of kidney disease induced by
a high-fat diet. J Am Soc Nephrol. 22:1846–1855. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hallows KR, Mount PF, Pastor-Soler NM and
Power DA: Role of the energy sensor AMP-activated protein kinase in
renal physiology and disease. Am JPhysiol Renal Physiol.
298:F1067–F1077. 2010. View Article : Google Scholar
|
|
12
|
Lv ZM, Liu Y, Zhang PJ, Xu J, Jia ZH, Wang
R and Wan Q: The role of AMPKα in high glucose-induced dysfunction
of cultured rat mesangial cells. Ren Fail. 34:616–621. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang S, Zhang M, Liang B, Xu J, Xie Z, Liu
C, Viollet B, Yan D and Zou MH: AMPKalpha2 deletion causes aberrant
expression and activation of NAD(P)H oxidase and consequent
endothelial dysfunction in vivo: Role of 26S proteasomes. Circ Res.
106:1117–1128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim MY, Lim JH, Youn HH, Hong YA, Yang KS,
Park HS, Chung S, Ko SH, Shin SJ, Choi BS, et al: Resveratrol
prevents renal lipotoxicity and inhibits mesangial cell
glucotoxicity in a manner dependent on the AMPK-Sirt1-PGC1α axis in
db/db mice. Diabetologia. 56:204–217. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Salminen A, Hyttinen JM and Kaarniranta K:
AMP-activated protein kinase inhibits NF-κB signaling and
inflammation: Impact on healthspan and lifespan. J Mol Med (Berl).
89:667–676. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee JH, Kim JH, Kim JS, Chang JW, Kim SB,
Park JS and Lee SK: AMP-activated protein kinase inhibits TGF-β-,
angiotensin II-, aldosterone, high glucose-, and albumin-induced
epithelial-mesenchymal transition. Am J Physiol Renal Physiol.
304:F686–F697. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raje N and Anderson K: Thalidomide - a
revival story. N Engl J Med. 341:1606–1609. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Asher C and Furnish T: Lenalidomide and
thalidomide in the treatment of chronic pain. Expert Opin Drug Saf.
12:367–374. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Amirshahrokhi K and Ghazi-Khansari M:
Thalidomide attenuates multiple low-dose streptozotocin-induced
diabetes in mice by inhibition of proinflammatory cytokines.
Cytokine. 60:522–527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao J, Wang H, Song T, Yang Y, Gu K, Ma
P, Zhang Z, Shen L, Liu J and Wang W: Thalidomide promotes morphine
efficacy and prevents morphine-induced tolerance in rats with
diabetic neuropathy. Neurochem Res. 41:3171–3180. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Behl T, Kaur I, Goel H and Kotwani A:
Significance of the antiangiogenic mechanisms of thalidomide in the
therapy of diabetic retinopathy. Vascul Pharmacol. 92:6–15. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim DH, Kim YJ, Chang SA, Lee HW, Kim HN,
Kim HK, Chang HJ, Sohn DW and Park YB: The protective effect of
thalidomide on left ventricular function in a rat model of diabetic
cardiomyopathy. Eur J Heart Fail. 12:1051–1060. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang H, Yang Y, Wang Y, Wang B and Li R:
Renal-protective effect of thalidomide in streptozotocin-induced
diabetic rats through anti-inflammatory pathway. Drug Des Devel
Ther. 12:89–98. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xu WW, Guan MP, Zheng ZJ, Gao F, Zeng YM,
Qin Y and Xue YM: Exendin-4 alleviateshigh glucose-induced rat
mesangial cell dysfunction throughthe AMPK pathway. Cell Physiol
Biochem. 33:423–432. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
García-García PM, Getino-Melián MA,
Domínguez-Pimentel V and Navarro-González JF: Inflammation in
diabetic kidney disease. World J Diabetes. 5:431–443. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Williams MD and Nadler JL: Inflammatory
mechanisms of diabetic complications. Curr Diab Rep. 7:242–248.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tesch GH: MCP-1/CCL2: A new diagnostic
marker and therapeutic target for progressive renal injury in
diabetic nephropathy. Am J Physiol Renal Physiol. 294:F697–F701.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morii T, Fujita H, Narita T, Shimotomai T,
Fujishima H, Yoshioka N, Imai H, Kakei M and Ito S: Association of
monocyte chemoattractant protein-1 with renal tubular damage in
diabetic nephropathy. J Diabetes Complications. 17:11–15. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng J, Encarnacion Diaz MM, Warner GM,
Gray CE, Nath KA and Grande JP: TGF-beta1 stimulates monocyte
chemoattractant protein-1 expression in mesangial cells through a
phosphodiesterase isoenzyme 4-dependent process. Am J Physiol Cell
Physiol. 289:C959–C970. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee MJ, Feliers D, Mariappan MM,
Sataranatarajan K, Mahimainathan L, Musi N, Foretz M, Viollet B,
Weinberg JM, Choudhury GG and Kasinath BS: A role for AMP-activated
protein kinase in diabetes-induced renal hypertrophy. Am J Physiol
Renal Physiol. 292:F617–F627. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao J, Miyamoto S, You YH and Sharma K:
AMP-activated protein kinase (AMPK) activation inhibits nuclear
translocation of Smad4 in mesangial cells and diabetic kidneys. Am
J Physiol Renal Physiol. 308:F1167–F1177. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dugan LL, You YH, Ali SS, Diamond-Stanic
M, Miyamoto S, DeCleves AE, Andreyev A, Quach T, Ly S, Shekhtman G,
et al: AMPK dysregulation promotes diabetes-related reduction of
superoxide and mitochondrial function. J Clin Invest.
123:4888–4899. 2013. View
Article : Google Scholar : PubMed/NCBI
|