Introduction
Acquired hemophilia A (AHA) is a clinically rare
coagulopathy that results in soft tissue and mucocutaneous
hemorrhage, and possible life-threatening bleeding (1). It has been reported that the annual
incidence rate of AHA is ~2 individuals per million, worldwide
(2). In the majority of cases,
excessive bleeding episodes are spontaneous at presentation
(2). The disorder presents without
personal or family history of bleeding, and has a relatively high
mortality rate, estimated at 9–22% (3).
To date, the pathogenesis of AHA has remained
unclear. Only ~50% of reported cases are typically associated with
autoimmune disorders, malignancy, adverse drug reactions and/or
various skin diseases (4). The
optimal hemostatic therapy is controversial due to low incidence.
The most common treatments for AHA with acquired factor VIII
(FVIII) deficiency are bleeding management, eradication of the
factor VIII inhibitor, treatment of underlying diseases, and
decreasing the risk of injuries that may cause iatrogenic bleeding
(2).
Herein, the present report documents a case of AHA
with acquired FVIII deficiency associated with intra-abdominal
hemorrhage, muscle hemorrhage and hemothorax 48 days after
premature delivery. The diagnosis process included imaging and
laboratory examinations. The patient was successfully diagnosed
with AHA and treated with an infusion of prothrombin complex to
prevent progressive bleeding and a glucocorticoid to eradicate the
factor VIII inhibitor. Therefore, it may be proposed that it is
important to differentiate AHA from another diseases, including
congenital hemophilia A with inhibitors and lupus anticoagulants
(LAs), which presents with spontaneous hemorrhaging or prolonged
activated partial thromboplastin time (aPTT). Also presented is a
review of AHA with regard to its epidemiology, associated risk
factors, disease course and recent research concerning its
management.
Case report
The current report was approved by the Ethics
Committee of the First Affiliated Hospital of Zhejiang Chinese
Medical University (Hangzhou, China). A 35-year-old woman who
presented with persistent fever and right lower back pain for 4
days, as well as dizziness, right leg pain and right abdominal pain
for 1 day, was admitted to the emergency department at the First
Affiliated Hospital of Zhejiang Chinese Medical University on
October 22, 2017. The patient had loss of sensation and diminished
range of motion in the right leg, and transient loss of
consciousness with hypotension. She had undergone childbirth 48
days prior to admission, and had no history of trauma or family
history of hemopathy. The patient did have a history of
appendicitis and no related surgeries.
On physical examination, abnormal vital signs
included a blood pressure of 89/58 mmHg (normal range, 90–140/60–90
mmHg) and temperature of 38°C. In a lung examination, respiration
was determined to be absent in the right lower lung. Shifting
dullness was noted by percussion of the abdomen. The patient had
severe pressure pain in the right lower abdomen, inner thigh and
back, as well as percussion pain in the kidney region; furthermore,
the right inner thigh appeared swollen. No peripheral edema or
hepatosplenomegaly was noted.
A pelvic ultrasound confirmed a postpartum uterus
and showed a suspicious teratoma in the left adnexal area, and a
routine blood examination revealed a white blood cell (WBC) count
of 10.1×109/l (normal range, 3.5–9.5×109/l),
and hemoglobin (Hb; 118 g/l; normal range, 115–150 g/l) and
platelet counts (240×109/l; normal range,
125–350×109/l) within the normal range, 3 days prior to
admission (Hangzhou First People's Hospital; Huangzhou, China). The
patient had no history of anticoagulation treatment or coagulopathy
and no signs of inflammation of the abdomen, respiratory tract or
urinary tract.
The patient underwent a series of examinations
following admission, including for WBC count
(17.9×109/l), neutrophil granulocyte rate (75.8%; normal
range, 40.0–75.0%), Hb (70 g/l; normal range, 115–150 g/l),
C-reactive protein (CRP; 73.09 mg/l; normal range, 1–8mg/l),
procalcitonin (PCT; 0.07 ng/ml; normal range, 0–0.05 ng/ml), aPTT
(68.4; normal range, 25.0–36.0 sec); D-dimer (1.37; normal range,
0–0.55 mg/l) and fibrinogen [FIB; 4.11 g/l; normal range, 2.00–4.00
g/l; with a normal international normalized ratio (INR; 0.94;
normal range, 0.8–1.2)]. Negative results were obtained for the
serum tumor marker test, the Rous test which detected the
metabolite of hemoglobin in urine to indicate that hemoglobinuria
had been present recently, the Coombs test, complement regulatory
protein cluster of differentiation (CD)55/CD59, human leucocyte
antigen antibodies, the antinuclear antibodies (ANCA) profile,
cytomegalovirus DNA, Epstein Barr virus DNA, virus immunoglobulin
(Ig)M antibody, anticardiolipin IgM and IgA, autoimmune hepatitis
antibody, the tuberculin purified protein derivative (PPD) test,
and rheumatoid factors. The patient's autoantibody (ANA) profile
was 1:320 (normal range: <1:100).
Right pleural effusion, abdominal effusion and a
dark liquid area in the posterior right abdomen and lumbar regions
were observed during bedside ultrasonic testing. Abdominal and
chest computed tomography (CT) revealed a markedly swollen right
lumbar muscle, exudative lesions of the soft tissues surrounding
the right groin, right thigh and right abdominal cavity,
retroperitoneal and pelvic effusion, and right massive pleural
effusion with incomplete expansion of the right lung (Fig. 1). Mild mitral valve reflux and
tricuspid valve regurgitation, high pulmonary artery systolic blood
pressure, and reduced diastolic left ventricular function were
observed on heart doppler ultrasonography (Fig. 2).
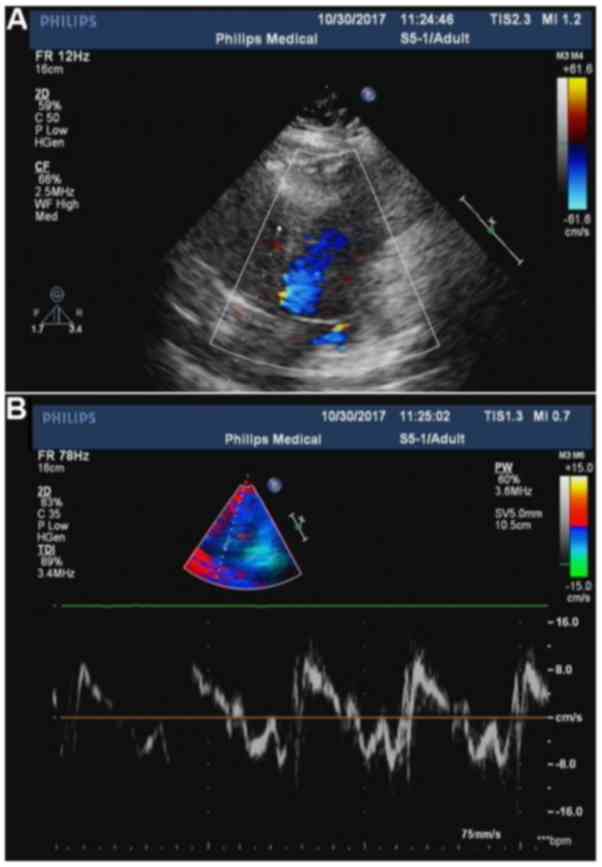 | Figure 2.(A) Color doppler ultrasonography
showing mild mitral valve reflux. (B) Tissue doppler imaging
showing tricuspid valve regurgitation, high pulmonary artery
systolic blood pressure and reduced diastolic left ventricular
function. The parameters of cardiac function were as follows: Right
ventricular internal dimension in diastole, 26.1 mm;
intraventricular septum in diastole, 7.6 mm; left ventricular
internal dimension-in diastole, 43.1 mm; left ventricular posterior
wall dimension, 7.6 mm; left ventricular internal dimension in
systole, 23.4 mm; heart rate, 84 bpm; aorta, 23.1 mm; left atrium,
32.5 mm; fractional shortening, 45.7%; ejection fraction,
77.4%). |
The following were observed by thromboelastography
(TEG): Reaction time (time from the start of the test to initial
fibrin formation), 35.8 min (normal range, 5.0–10.0 min),
coagulation time (time required for clot formation), 15.3 min
(normal range, 1.0–3.0 min), angle (measurement of clot growth),
35.2 (normal range, 53.0–72.0°), maximum amplitude (measurement of
clot strength), 44.5 mm (normal range, 50.0–70.0 mm) and
coagulation index, −28.8 (normal range, −3.0–3.0; Fig. 3). Markedly decreased FVIII activity
(12.6%; normal range, 60.0–150.0%), decreased FVIII procoagulant
activity (FVIII: C; 0.8%; normal range, 50.0–150.0%) and a
high-titer of the FVIII inhibitor [7.4 Bethesda units (BU)/ml;
normal range, 0–0.6 BU/ml] was observed in a coagulation factor
assay.
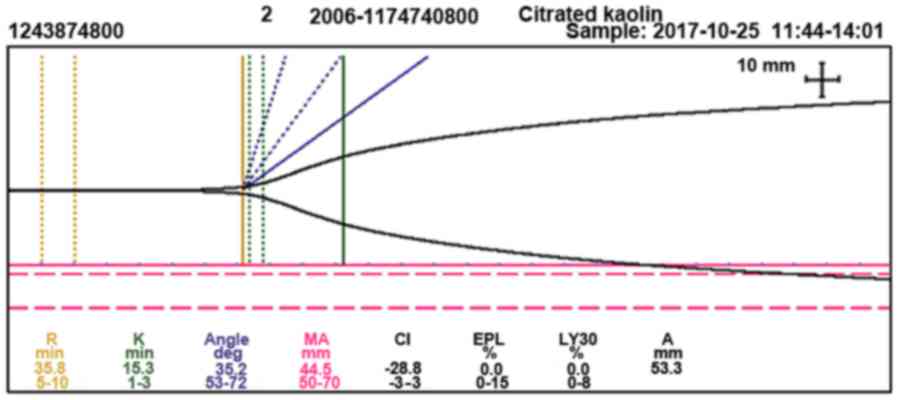 | Figure 3.Thromboelastography showing a
prolonged R-time (35.8 min) and K-time (15.3 min), a narrowed angle
(35.2°), decreased MA (44.5 mm), decreased CI (−28.8) and normal
clot lysis, which is reported in terms of EPL (0.0%) and LY30%
(0.0%). R, reaction time; K, coagulation time; deg, degree; MA,
maximum amplitude; CI, coagulation index; EPL, estimated percent
lysis; LY30%, percent clot lysis at 30 min after maximum amplitude;
A, amplitude. |
Collectively these examinations indicated that the
patient had AHA. Antishock and coagulation factor replacement were
provided to stop the bleeding. A transfusion of red blood cells,
plasma, prothrombin complex (PCC) and vitamin K was initiated
during the first 4 days of hospitalization. Due to concerns about
the patient's high WBC count and CRP and PCT levels, the antibiotic
meropenem was administered at 1.0 g every 12 h, to prevent
potential infections. Following a suspected diagnosis of AHA, the
patient was treated with intravenous human coagulation FVIII (1,600
IU twice daily), dexamethasone (5 mg/day) and intravenous Ig (10
g/day). Thoracic drainage for 7 days relieved the chest congestion,
which was determined to be exudative bloody pleural effusion, based
on cell counting and classification of the pleural effusion under
the microscope, as well as the Rivalta test (color, red; Rivalta
test, positive; WBC, 600.0 µl; neutrophil percentage, 46.0%;
lymphocytes percentage, 12.0%; mesothelial cells percentage,
42.0%). One week later, the patient experienced pain relief and
normal body temperature was recovered; in addition, the Hb levels
had risen (85 g/l; normal range, 115–150 g/l) and the extensive
subcutaneous ecchymoses of the right thigh and lower back had
disappeared. Therefore, the glucocorticoid treatment was changed to
oral prednisone, 20 mg three times a day, and then to oral
prednisone, 15 mg three times a day, 2 weeks later. Concurrently,
the patient was also administered calcitriol soft capsules (R.P.
Scherer GmbH & Co. KG, Eberbach, Germany), 1 capsule (0.25 µg)
twice a day, and calcium carbonate and vitamin D3 tablets (Wyeth
pharmaceuticals, Inc., Collegeville, PA, USA), 1 pill (calcium
carbonate 1.5 g and vitamin D3 125 IU) twice a day, to prevent
osteoporosis due to the glucocorticoid. At 1 month after treatment,
the pain and subcutaneous ecchymosis in the patient's right side
were relieved, a chest CT scan showed clearing of the exudative
lesions, and laboratory values gradually recovered, including those
of a routine blood examination, aPTT, FIB and D-dimer levels, and
the level of FVIII inhibitor was reduced (0 BU/ml).
The patient was discharged on November 17, 2017. Her
discharge medication consisted of prednisone, 10 mg three time a
day, calcitriol soft capsules, 1 capsule twice a day, and calcium
carbonate and vitamin D3 tablets, 1 pill twice a day. Moreover, we
suggested weekly follow-ups. There was no indication of relapse at
the 2-month follow-up.
Discussion
In the present case, a rapid accurate diagnosis was
a prerequisite for successful treatment. Patients with polyserous
effusions typically visit an oncologist, surgeon or respiratory
physician (5–7). In the absence of trauma history and a
family history of bleeding disorders in the present case, as well
as negative results on tumor protein expression analysis and
tuberculin PPD testing, genetic diseases, malignancy and
tuberculosis were excluded. Therefore, hematopoietic diseases were
considered. The patient's aPTT was elevated with normal INR and
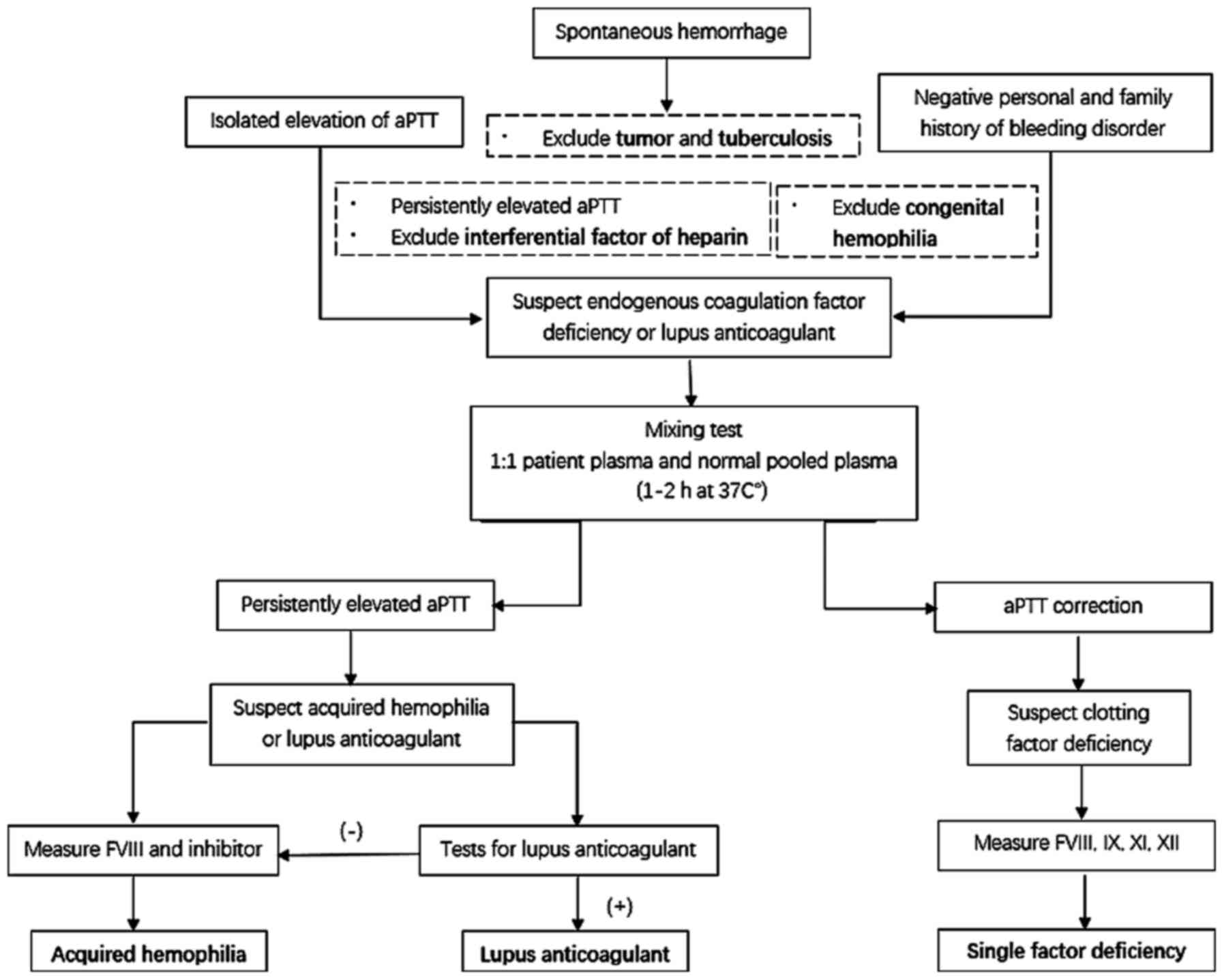
normal prothrombin time. We suspected endogenous coagulation
dysfunction or LA. The lack of aPTT correction following a mixing
test was cause for suspected AH or LA.
The TEG analysis indicated a coagulation factor
abnormality. Following consideration of the above results along
with the markedly decreased FVIII activity, it was inferred that
the patient's pathology was associated with FVIII deficiency.
Moreover, the presence of FVIII inhibitor verified the suspected
AHA (Fig. 4).
It is necessary to differentiate AHA from congenital
hemophilia A with inhibitors, since the latter indicates a personal
and family history of bleeding disorders since childhood and
exhibits clinical characteristics that include muscle and joint
spontaneous hemorrhage and joint deformity; moreover, it is
consistent with the X-linked recessive inheritance law (8). The same antibody produced by a
hemophilia A patient may completely inactivate FVIII without
residual FVIII: C (2). Therefore,
the hemostatic effect is not produced following infusion of FVIII
preparations at the same dose that may have been effective
previously (9).
In addition, LA presents with a persistent prolonged
aPTT, as does AHA, and may lead to the false appearance of reduced
coagulation factors in vitro due to its inhibitory effect on
phospholipids (10). In LA,
prolonged aPTT cannot be corrected by using normal plasma; however,
it can be shortened and corrected by the supplementation of
exogenous phospholipids (11). This
can be proven more definitively through a variety of
phospholipid-dependency experiments and by performing the Dilute
Russell's viper venom time test. FVIII autoantibodies and LA may
coexist in the same patient (11,12). For
complicated cases, ELISA may be used to identify FVIII inhibitors
in LA (12). An aPTT reagent, which
is not sensitive to LA, can be used to eliminate its effect on
coagulation (2). Clinically,
patients with LA primarily manifest with thrombosis events, and
bleeding is rarely observed (12,13).
Although there is reportedly no significant
difference in the incidence of AHA between the sexes, it is
predominantly a disease of the elderly aged ≥60 years (7). However, it can also be associated with
pregnancy and autoimmune disease in younger groups (aged 20–30
years) (2,14). Most older patients with AHA tend to
be women, while most younger patients tend to be men (14,15).
Reports on postpartum AHA are rare. In a previous
study, ~10% of AHA cases were associated with pregnancy (16). The hemorrhagic symptoms in female
patients are commonly present between 1 and 4 months after
parturition (17,18).
AHA is often associated with autoimmune disease,
including rheumatoid arthritis, systemic lupus erythematosus and
myasthenia gravis (2). The incidence
of AHA associated with autoimmune disorders has been demonstrated
to be as high as 20% in all AHA patients, worldwide (19). High FVIII inhibitor titers often
appear in patients with autoimmune disorders as well as pregnant
individuals (2). However, in
postpartum subjects, the hemorrhagic potential is often low and the
inhibitors spontaneously disappear in almost all of these patients,
with titers of inhibitors lower than those of patients with an
autoimmune disorder (17).
Furthermore, it may be difficult to achieve inhibitor eradication
in patients with high titers (≥5 BU/ml), even with aggressive
immunosuppressive therapy (2). The
pathogenesis may be that carrying a fetus could induce the risk of
fatal bleeding since it induces the risk of diaplacental transition
and the development of postpartum inhibitors in pregnant AHA
patients (2). Therefore, the
presence of an autoimmune disease should be considered in this
patient and with continued monitoring of ANA and ANCA profiles to
detect autoimmune pathogens.
AHA is an important disorder clinically and
economically, and often unrecognized or misdiagnosed, which may be
reason for its relatively high mortality rate, estimated to be
~30%, worldwide (16,20). Therefore, aggressive treatment is
recommended to decrease the mortality rate in AHA (14). The main goals of AHA management are
to control and prevent bleeding, eradicate the FVIII inhibitor and
treat the underlying disease. The first-line treatment for severe
bleeding episodes is administration of bypassing agents, including
activated prothrombin complex concentrates (APCCs) containing
factors II, VII, IX, X and VIIa, human FVIII and recombinant
activated factor VII (5). In the
present case, FVII was not administered because of its high cost
and the beneficial therapeutic effect that was provided by
administration of APCC and FVIII. Agents used in immunosuppressive
therapy for suppressing the FVIII inhibitor include long-term
immunosuppressive agents, consisting of corticosteroids alone or in
combination with azathioprine, cyclosporine and antineoplastic
agents, including cyclophosphamide, mercaptopurine and vincristine
(2,6). The use of intravenous immunoglobulin in
multiple treatment sessions is another eradication strategy;
however, its effects remain unclear.
Infectious diseases, including pneumonia and sepsis,
are responsible for ~50% of AHA-associated mortalities (21). Therefore, sufficient attention to the
prevention and early detection of infectious diseases is warranted
when aggressive and prolonged immune suppression therapy is
provided (2). For these reasons,
antimicrobial agents were also prescribed presently.
In conclusion, the current report describes, to our
knowledge, the first reported case of AHA with a possible
autoimmune disorder during the postpartum period in a 35-year-old
female who presented with polyserous bloody effusions. Awareness of
the correct diagnosis of AHA is necessary as it is a curable
disease, although it can be life threatening. It was also
highlighted how timely treatment can be successful and lifesaving.
Corticosteroids remain the most commonly prescribed
immunosuppressive therapy treatment for AHA patients, in addition
to bleeding management and treatment of the underlying disease.
Acknowledgements
The authors would like to thank Editage for the
scientific editing of the manuscript, and the staff at the
Departments of Hematology and Emergency, The First Affiliated
Hospital of Zhejiang Chinese Medical University for their help in
the management of this patient.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81503522)
and Zhejiang Natural Science Foundation (grant no.
LQ15H270004).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LX and ZZ made substantial contributions to
conception and design and revised the manuscript critically. XZ, JC
and XH made the contributions to acquisiton of data and wrote the
draft. YT, NZ and LW interpreted the results and were involved in
drafting the manuscript and revising it critically. All authors
have read and given final approval of version to be published and
agreed to be accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
This case was approved by the Ethics Committee of
the First Affiliated Hospital of Zhejiang Chinese Medical
University.
Patient consent for publication
Authors have disclosed to this patient that
personally identifiable material would be available via the
Internet as well as in print after publication. Written informed
consent was obtained from the patient under these conditions.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
FVIII
|
factor VIII
|
|
AHA
|
acquired hemophilia A
|
|
WBC
|
white blood cell
|
|
Hb
|
hemoglobin
|
|
CRP
|
C-reactive protein
|
|
PCT
|
procalcitonin
|
|
aPTT
|
activated partial thromboplastin
time
|
|
FIB
|
fibrinogen
|
|
INR
|
international normalized ratio
|
|
CD
|
cluster of differentiation
|
|
ANCA
|
antinuclear antibodies
|
|
IgM/IgA
|
immunoglobulin M/A
|
|
PPD
|
purified protein derivative
|
|
ANA
|
autoantibody
|
|
CT
|
computed tomography
|
|
TEG
|
thromboelastography
|
|
BU
|
Bethesda units
|
|
APCCs
|
activated prothrombin complex
concentrates
|
|
LA
|
lupus anticoagulant
|
References
|
1
|
Vautier M, de Boysson H, Creveuil C,
Repesse Y, Borel-Derlon A, Troussard X, Damaj GL, Bienvenu B,
Gautier P and Aouba A: Influence of factor VIII level and its
inhibitor titer on the therapeutic response to corticosteroids
alone in the management of acquired hemophilia. Medicine
(Baltimore). 95:e52322016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sakurai Y and Takeda T: Acquired
hemophilia A: A frequently overlooked autoimmune hemorrhagic
disorder. J Immunol Res. 2014:3206742014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Collins P, Baudo F, Huth-Kühne A,
Ingerslev J, Kessler CM, Castellano MEM, Shima M, St-Louis J and
Lévesque H: Research article Consensus recommendations for the
diagnosis and treatment of acquired hemophilia A. BMC Research
Notes Notes Research Notes. 3(161)2010.
|
|
4
|
Aljasser MI, Sladden C, Crawford RI and Au
S: Bullous pemphigoid associated with acquired hemophilia A: A rare
association of autoimmune disease. J Cutan Med Surg. 18:123–126.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Olteanu M, Niţu M and Golli A:
Tuberculosis mesenteric adenopathy and polyserositis. Rom J Morphol
Embryol. 53:835–840. 2012.PubMed/NCBI
|
|
6
|
Kruse-Jarres R, Kempton CL, Baudo F,
Collins PW, Knoebl P, Leissinger CA, Tiede A and Kessler CM:
Acquired hemophilia A: Updated review of evidence and treatment
guidance. Am J Hematol. 92:695–705. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bitting RL, Bent S, Li Y and Kohlwes J:
The prognosis and treatment of acquired hemophilia: A systematic
review and meta-analysis. Blood Coagul Fibrinolysis. 20:517–523.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chinese society of hematology: Chinese
expert consensus on the diagnosis and treatment of acquired
hemophilia A. Zhonghua Xue Ye Xue Za Zhi. 35:575–576. 2014.(In
Chinese). PubMed/NCBI
|
|
9
|
Coppola A, Franchini M, Castaman G,
Santagostino E, Santoro C, Santoro RC, Morfini M, Minno GD and
Rocino A; and on behalf of the AICE ad hoc Working Group, :
Treatment regimens with bypassing agents in patients with
hemophilia A and inhibitors: A survey from the Italian association
of hemophilia centers (AICE). Semin Thromb Hemost. 44:551–560.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brandt JT, Barna LK and Triplett DA:
Laboratory identification of lupus anticoagulants: Results of the
second international workshop for identification of lupus
anticoagulants. Thromb Haemost. 74:1597–1603. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumano O, Amiral J, Dunois C, Peyrafitte M
and Moore GW: Paired APTTs of low and high lupus anticoagulant
sensitivity permit distinction from other abnormalities and achieve
good lupus anticoagulant detection rates in conjunction with dRVVT.
Int J Lab Hematol. 24:2018.
|
|
12
|
Thrombosis and Hemostasis Group, Chinese
Society of Hematology, Chinese Medical Association: Chinese expert
consensus on the diagnosis and treatment of acquired hemophilia A
(Chinese). Chin J Hematol. 35:575–576. 2014.
|
|
13
|
Greaves M, Cohen H, MacHin SJ and Mackie
I: Guidelines on the investigation and management of the
antiphospholipid syndrome. BrJ Haematol. 109:704–715. 2000.
View Article : Google Scholar
|
|
14
|
Theodossiades G, Tsevrenis V, Nomikou E,
Dadiotis L and Kontopoulou-Griva L: Surgery-associated acquired
hemophilia A. Ann Hematol. 80:691–693. 2011.
|
|
15
|
Bouvry P and Recloux P: Acquired
hemophilia. Haematologica. 79:550–556. 1994.PubMed/NCBI
|
|
16
|
Sborov DW and Rodgers GM: Acquired
hemophilia A: A current review of autoantibody disease. Clinical
Advances in Hematol Oncol. 10:19–27. 2012.
|
|
17
|
Delgado J, Jimenez-Yuste V,
Hernandez-Navarro F and Villar A: Acquired haemophilia: Review and
meta-analysis focused on therapy and prognostic factors. Br J
Haematol. 121:21–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Collins PW and Percy CL: Advances in the
understanding of acquired haemophilia A: Implications for clinical
practice. Br J Haematol. 148:183–194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shetty S, Bhave M and Ghosh K: Acquired
hemophilia A: Diagnosis, aetiology, clinical spectrum and treatment
options. Autoimmun Rev. 10:311–316. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Federici AB, Budde U, Castaman G, Rand JH
and Tiede A: Current diagnostic and therapeutic approaches to
patients with acquired von Willebrand syndrome: A 2013 update.
Semin Thromb Hemost. 39:191–201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tanaka I, Amano K, Taki M, Oka T, Sakai M,
Shirahata A, Takata N, Takamatsu J, Taketani H, Hanabusa H, et al:
A 3-year consecutive survey on current status of acquired
inhibitors against coagulation factors in Japan: Analysis of
prognostic factors. Japanese J Thromb Hemost. 19:140–153. 2008.
View Article : Google Scholar
|