Introduction
Pleuroparenchymal fibroelastosis (PPFE) is a rare
interstitial lung disease (1), first
described in 1992 by Amitani et al (2) as ‘upper lobe pulmonary fibrosis’. PPFE
is histologically characterized by a thickened, fibrotic visceral
pleura with subpleural parenchymal fibrosis and elastosis,
predominantly of the upper lobes. Although an increased number of
PPFE cases have been reported, the characteristics of this disease
have not been well described, which may lead to misdiagnosis. The
present study reports on the case of a patient who presented with
extensive unilateral lung abnormalities following autologous
hematopoietic stem cell transplantation (HSCT) and determined the
key characteristics, with the aim of helping physicians to
distinguish PPFE from chronic infectious diseases.
Case presentation
A 34-year-old male patient was admitted to the
China-Japan Friendship Hospital (Beijing, China) in May 2017 due to
experiencing progressive cough and exertional dyspnea for 9 years.
The patient had been diagnosed with Hodgkin's lymphoma in 2011, and
received chemotherapy (including adriamycin, bleomycin, vincristine
and dacarbazine) and radiotherapy, followed by autologous HSCT 2
years later. The initial chest radiography after the
transplantation was normal; however, 10 years prior to admission to
our hospital, a follow-up chest computed tomography (CT) revealed a
small right-sided pneumothorax, albeit without symptoms. At 9 years
prior to admission, the patient had developed a progressive cough
and exertional dyspnea, and chest CT revealed the presence of an
exudative lesion in the lower right lung. A bronchoscopy and
bronchoalveolar lavage (BAL) were performed 5 years prior to
admission. Acid-fast bacilli were detected in the BAL fluid smear.
The patient was then diagnosed with tuberculosis and received
treatment with isoniazide, rifampicin, pyrazinamide and
streptomycin for a total of 2 months, followed by isoniazide,
rifampicin and streptomycin for a further 16 months. However, the
symptoms slowly progressed. A biopsy of the right lung performed in
another hospital in 2016 revealed collagenous fibers and a certain
amount of normal alveolar tissue (Fig.
1). The patient was diagnosed with tuberculosis-associated lung
destruction. The patient was a non-smoker and reported no known
exposure to environmental allergens or asbestos.
On physical examination the patient was cachectic,
with a body mass index of 18.5 kg/m2 and had a flattened
thoracic cage. The heart rate was >120 beats/min and the
respiratory rate was 30 breaths/min. The breath sounds were
decreased at the bottom of the right lung and arterial blood gas
analysis revealed hypoxia. Pulmonary function tests revealed
restrictive ventilation dysfunction and decreased diffusion
capacity: The forced vital capacity (FVC) was 1.15 l (21.3% of
predicted) and the ratio of the forced expiratory volume in 1 sec
to the FVC was 95.14%; the total lung capacity was 2.72 l (35.7% of
predicted) and the diffusion capacity of carbon monoxide was 3.35
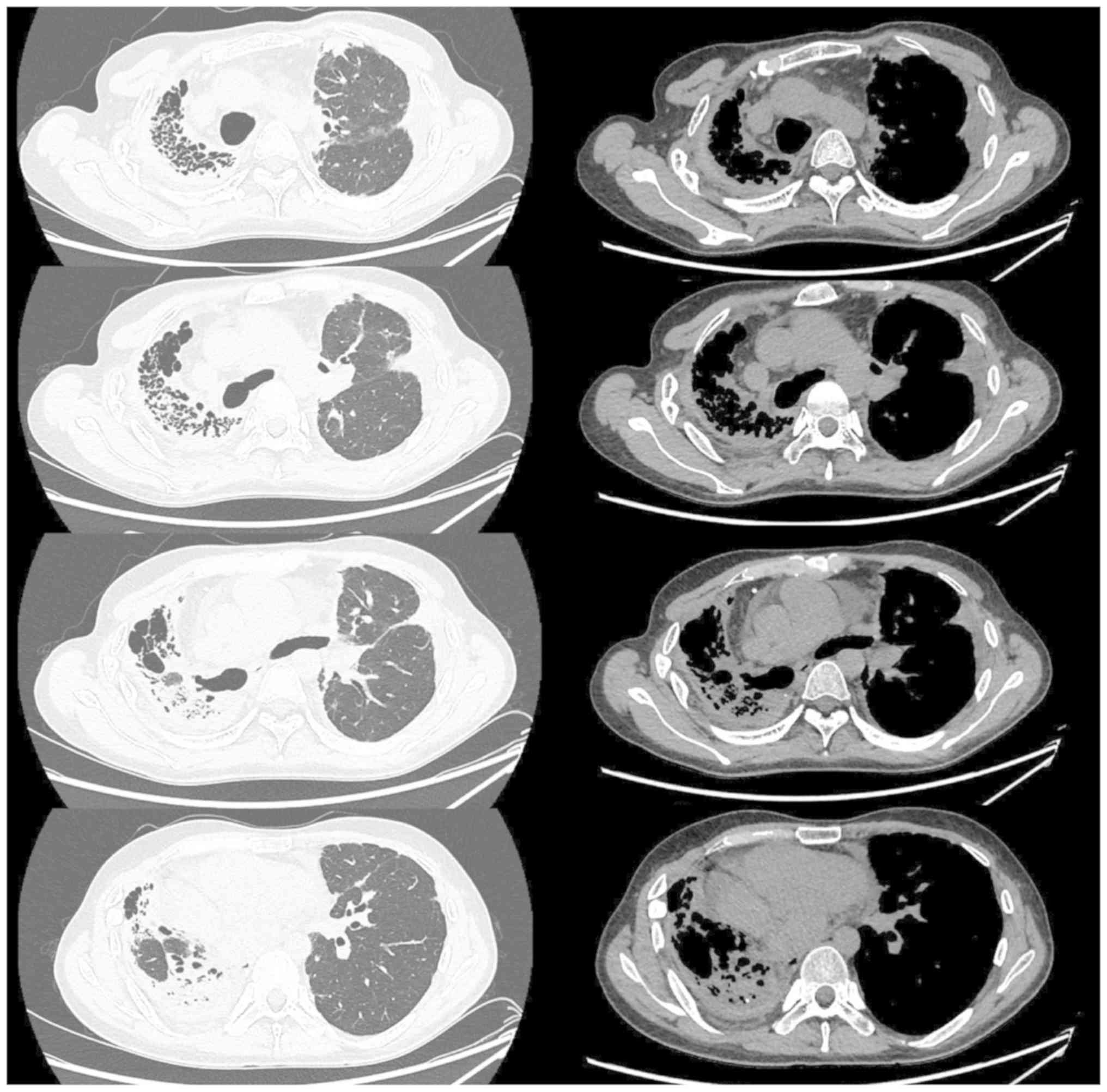
mmol/min/kPa (27.5% of predicted). Serial chest CTs performed at
the time of admission to our hospital revealed a gradually
worsening diffuse pleural thickening, dense subpleural
opacification and volume loss associated with evidence of fibrosis
in the right lung (Fig. 2).
Bronchoscopy was repeated at our hospital, with the BAL examination
revealing 5.0% macrophages, 17.5% lymphocytes, 76% neutrophils and
1.5% eosinophils. The microbiological and cytological examinations
were negative. Percutaneous lung puncture biopsy revealed a
thickened pleura, consisting of large amounts of collagen and
elastic fibers, coexisting with subpleural intra-alveolar fibrosis
and alveolar septal elastosis, without inflammatory infiltrates
(Fig. 3).
Based on the medical history, clinical
manifestations, imaging and histological findings, the patient was
diagnosed with PPFE secondary to HSCT and eventually succumbed to
respiratory failure and infection while waiting for a lung
transplant in August 2017.
Discussion
PPFE is a rare form of interstitial lung disease
characterized by elastic fibrosis involving the pleura and
subpleural parenchyma. Since PPFE was first described by Amitani
et al (2), >100 cases have
been reported to date (3–9). PPFE may be either idiopathic or occur
secondary to lung transplantation (10), marrow transplantation (3,11) or
HSCT (12), representing a rare late
post-transplantation complication.
The correlation between PPFE and transplantation was
first reported by von der Thüsen et al (11), who described PPFE secondary to bone
marrow transplantation. A study by Mariani et al (12) retrospectively reviewed
high-resolution computed tomography images from 53 lung transplant
recipients and 700 HSCT recipients, revealing that the prevalence
of PPFE was 7.54% [95% confidence interval (CI): 0.43–14.6%] among
lung transplant recipients and 0.28% (95% CI: 0.00–0.68%) among
HSCT recipients. In that retrospective study, patients with
secondary PPFE developed fibrosis within 2–13 years (mean, 5.3
years) post-transplantation. The case of the present study
developed pleural thickening and fibrosis 9 years after HSCT, which
is within the time window reported by previous studies. The
mechanism by which transplantation leads to PPFE has remained to be
fully elucidated. Possible causes may include reactions to
chemotherapy or radiotherapy, or graft-versus-host disease (GVHD)
(13). GVHD may be a likely cause,
as the majority of the reported cases occurred after HSCT. However,
certain patients develop PPFE after autologous bone marrow
transplantation, lung transplantation or chemotherapy alone. One
case developed post-HSCT PPFE and the surgical specimen exhibited
characteristics of PPFE, without any evidence of GVHD (i.e. no
lymphocytic inflammation or eosinophilic scarring suggestive of
GVHD or obliterative bronchiolitis) (14). In this case, immunosuppressive
therapy did not improve the pulmonary function (14). Those results suggest an association
of PPFE with the conditioning treatment for HSCT rather than with
GVHD. Therefore, PPFE after HSCT is a heterogeneous condition, with
the contribution of GVHD to the development of PPFE differing
across cases. According to certain experts, in addition to lung
collapse and fibrosis resulting from constrictive bronchiolitis
obliterans, PPFE may be a consequence of persistent intra-alveolar
organizing pneumonia (11). By
histological analysis of biopsy specimens, certain studies
demonstrated that diffuse alveolar damage preceded the development
of PPFE in lung transplant as well as in HSCT recipients,
suggesting that PPFE may represent a late complication associated
with multiple factors (drugs, radiation, infection and
cell-mediated immune reaction) that results in acute lung
injury/diffuse alveolar damage. PPFE has also been described as a
pulmonary complication following chemotherapy (15) or radiotherapy (16). The present patient received
chemotherapy and radiotherapy at another hospital 17 years ago.
Unfortunately, no information on the dosage of chemotherapy and
radiotherapy was available, which is a limitation of the present
study.
The case of the present study was diagnosed with
tuberculosis-induced lung destruction prior to presenting at our
hospital, and had received regular anti-tuberculosis therapy for 18
months, although the lesion on the right lung continued to
progress. Possible reasons included non-tuberculosis mycobacterial
infection rather than tuberculosis, and other diseases leading to
structural damage of the lung. Considering that pneumothorax, which
is a known clinical characteristic of PPFE, had occurred years
prior to the diagnosis of tuberculosis, and that a significant
deterioration with diffuse bilateral pleural thickening, albeit
without inflammatory infiltrates, was observed on biopsy, the
diagnosis of PPFE was favored over that of an infectious disease.
Recurrent pulmonary infections may be an important risk factor for
the progression of PPFE. Pulmonary infectious diseases caused by
Mycobacterium avium-intracellulare complex (17), Aspergillus (18,19) or
Cytomegalovirus (12) have
been reported in patients with PPFE. These infectious diseases may
coexist, but the association between these infections and PPFE has
not yet been established. This calls for further investigation into
whether the pathology of PPFE is induced by these infections, or
whether PPFE favors the growth of these infectious pathogens.
The patient underwent a consecutive series of chest
CT scans during follow-up over the 9 years prior to admission to
our hospital. However, the destruction of the lung parenchyma was
attributed to tuberculosis, even after a biopsy of the right lung.
A better awareness of the clinical, radiological and histological
characteristics of PPFE will help physicians distinguish between
this disease and chronic infections. These characteristics include
typical risk factors (lung transplantation, bone marrow
transplantation, HSCT, chemotherapy and inhalational exposure to
aluminosilicate), slow progression, platythorax and marked
thickening of the pleura with elastic fibers and dense
collagen.
The prognosis for PPFE remains poor, with variable
progression (18,20). The clinical course was reported to be
progressive in a number of PPFE patients, despite aggressive
treatment with corticosteroids, immunosuppressants or pirfenidone
(19,21). Lung transplantation has been applied
as a treatment in several cases of end-stage PPFE, but long-term
outcome data are currently unavailable (21–23).
Acknowledgements
The authors would like to thank Dr Elisabetta A.
Renzoni (Interstitial Lung Disease Unit, Royal Brompton Hospital,
Imperial College, London, UK), and Dr Francesca Mariani and Dr
Maurizio Zompatori (Department of Radiology, S. Orsola-Malpighi
Hospital, Bologna, Italy) for their pathological and radiological
advice for the case.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81600036), the National
Natural Science Foundation of China (grant no. 81570049) and the
National Key Research and Development Program of China (grant no.
2016YFC0905600).
Availability of data and materials
All the datasets generated and analyzed in the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
SZ and WX analyzed and interpreted the patient data
and were major contributors in writing the manuscript. ZW and YT
wrote the original medical record of the patient. JD performed the
histological examination of the biopsy tissue. ZZ revised the
manuscript. All authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Informed consent has been obtained from the patient
regarding the publication of the case details and any associated
images.
Competing interests
The authors declare that they have no competing
interests to disclose.
References
|
1
|
Travis WD, Costabel U, Hansell DM, King TE
Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU,
et al: ATS/ERS committee on idiopathic interstitial pneumonias. An
official american thoracic society/European respiratory society
statement: Update of the international multidisciplinary
classification of the idiopathic interstitial pneumonias. Am J
Respir Crit Care Med. 188:733–748. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Amitani R, Niimi A and Kuse F: Idiopathic
pulmonary upper lobe fibrosis (IPUF). Kokyu. 11:693–699. 1992.
|
|
3
|
Fujikura Y, Kanoh S, Kouzaki Y, Hara Y,
Matsubara O and Kawana A: Pleuroparenchymal broelastosis as a
series of airway complications associated with chronic
graft-versus-host disease following allogeneic bone marrow
transplantation. Intern Med. 53:43–46. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng SK and Chuah KL: Pleuroparenchymal
fibroelastosis of the lung: A review. Arch Pathol Lab Med.
140:849–853. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoshida Y, Nagata N, Tsuruta N, Kitasato
Y, Wakamatsu K, Yoshimi M, Ishii H, Hirota T, Hamada N, Fujita M,
et al: Heterogeneous clinical features in patients with pulmonary
fibrosis showing histology of pleuroparenchymal fibroelastosis.
Respir Investig. 54:162–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rosenbaum JN, Butt YM, Johnson KA, Meyer
K, Batra K, Kanne JP and Torrealba JR: Pleuroparenchymal
fibroelastosis: A pattern of chronic lung injury. Hum Pathol.
46:137–146. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hirota T, Yoshida Y, Kitasato Y, Yoshimi
M, Koga T, Tsuruta N, Minami M, Harada T, Ishii H, Fujita M, et al:
Histological evolution of pleuroparenchymal fibroelastosis.
Histopathology. 66:545–554. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Enomoto N, Kusagaya H, Oyama Y, Kono M,
Kaida Y, Kuroishi S, Hashimoto D, Fujisawa T, Yokomura K, Inui N,
et al: Quantitative analysis of lung elastic fibers in idiopathic
pleuroparenchymal fibroelastosis (IPPFE): Comparison of clinical,
radiological, and pathological findings with those of idiopathic
pulmonary fibrosis (IPF). BMC Pulm Med. 14:912014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kusagaya H, Nakamura Y, Kono M, Kaida Y,
Kuroishi S, Enomoto N, Fujisawa T, Koshimizu N, Yokomura K, Inui N,
et al: Idiopathic pleuroparenchymal fibroelastosis: Consideration
of a clinicopathological entity in a series of Japanese patients.
BMC Pulm Med. 12:722012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hirota T, Fujita M, Matsumoto T, Higuchi
T, Shiraishi T, Minami M, Okumura M, Nabeshima K and Watanabe K:
Pleuroparenchymal fibroelastosis as a manifestation of chronic lung
rejection? Eur Respir J. 41:243–245. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
von der Thüsen JH, Hansell DM, Tominaga M,
Veys PA, Ashworth MT, Owens CM and Nicholson AG: Pleuroparenchymal
fibroelastosis in patients with pulmonary disease secondary to bone
marrow transplantation. Mod Pathol. 24:1633–1639. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mariani F, Gatti B, Rocca A, Bonifazi F,
Cavazza A, Fanti S, Tomassetti S, Piciucchi S, Poletti V and
Zompatori M: Pleuroparenchymal fibroelastosis: The prevalence of
secondary forms in hematopoietic stem cell and lung transplantation
recipients. Diagn Interv Radiol. 22:400–406. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Takeuchi Y, Miyagawa-Hayashino A, Chen F,
Kubo T, Handa T, Date H and Haga H: Pleuroparenchymal
fibroelastosis and non-specific interstitial pneumonia: Frequent
pulmonary sequelae of haematopoietic stem cell transplantation.
Histopathology. 66:536–544. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Okimoto T, Tsubata Y, Hamaguchi M, Sutani
A, Hamaguchi S and Isobe T: Pleuroparenchymal fibroelastosis after
haematopoietic stem cell transplantation without graft-versus-host
disease findings. Respirol Case Rep. 6:e002982018.PubMed/NCBI
|
|
15
|
Beynat-Mouterde C, Beltramo G, Lezmi G,
Pernet D, Camus C, Fanton A, Foucher P, Cottin V and Bonniaud P:
Pleuroparenchymal fibroelastosis as a late complication of
chemotherapy agents. Eur Respir J. 44:523–527. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Watanabe K: Pleuroparenchymal
fibroelastosis: Its clinical characteristics. Curr Respir Med Rev.
9:299–237. 2013.PubMed/NCBI
|
|
17
|
Watanabe K, Nagata N, Kitasato Y,
Wakamatsu K, Nabeshima K, Harada T, Hirota T, Shiraishi M and
Fujita M: Rapid decrease in forced vital capacity in patients with
idiopathic pulmonary upper lobe fibrosis. Respir Investig.
50:88–97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Piciucchi S, Tomassetti S, Casoni G,
Sverzellati N, Carloni A, Dubini A, Gavelli G, Cavazza A, Chilosi M
and Poletti V: High resolution CT and histological findings in
idiopathic pleuroparenchymal fibroelastosis: Teatures and
differential diagnosis. Respir Res. 12:1112011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reddy TL, Tominaga M, Hansell DM, von der
Thusen J, Rassl D, Parfrey H, Guy S, Twentyman O, Rice A, Maher TM,
et al: Pleuroparenchymal fibroelastosis: A spectrum of
histopathological and imaging phenotypes. Eur Respir J. 40:377–385.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bonifazi M, Montero MA and Renzoni EA:
Idiopathic pleuroparenchymal fibroelastosis. Curr Pulmonol Rep.
6:9–15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang H, Feng R, Li S, Wu B, Xu K, Xu Z
and Chen J: A CARE-compliant case report: Lung transplantation for
a Chinese young man with idiopathic pleuroparenchymal
fibroelastosis. Medicine (Baltimore). 96:e69002017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen F, Matsubara K, Miyagawa-Hayashino A,
Tada K, Handa T, Yamada T, Sato M, Aoyama A and Date H: Lung
transplantation for pleuroparenchymal fibroelastosis after
chemotherapy. Ann Thorac Surg. 98:e115–e117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hata A, Nakajima T, Yoshida S, Kinoshita
T, Terada J, Tatsumi K, Matsumiya G, Date H and Yoshino I: Living
donor lung transplantation for pleuroparenchymal fibroelastosis.
Ann Thorac Surg. 101:1970–1972. 2016. View Article : Google Scholar : PubMed/NCBI
|