Introduction
Unexplained recurrent miscarriage (RM) is defined as
≥3 consecutive idiopathic miscarriages prior to the gestational age
of 12 weeks of pregnancy. RM affects 1–2% of females attempting to
conceive and represents a considerable challenge for physicians
(1,2). RM may be explained by genetic
abnormalities, maternal immunological causes and endocrine
dysfunction only in a minority of cases, while the majority of
cases are diagnosed as unexplained RM (3). The vagina harbors different species of
bacteria in variable quantities and relative proportions that have
important effects on the health of women (4). A healthy vaginal flora, dominated by
Lactobacillus species, has an important role in the
protection against genital infections, which are considered as a
common cause of miscarriage (5). A
change in the bacterial flora of the vagina is represented by a
decrease of Lactobacillus and overgrowth of several
anaerobic or facultative bacteria, including Mobiluncus
species, Prevotella species, Gardnerella vaginalis
and genital Mycoplasm: Mycoplasma hominis and
Ureaplasma urealyticum (6).
At present, bacterial vaginosis is thought to be mainly caused by
vaginal dysbacteriosis, which may lead to premature birth,
premature rupture of membranes, low birth weight at premature
birth, RM, chorioamnionitis and a series of diseases (7).
Advances in sequencing technologies and analytical
methods have enabled the exploration of the human microbiota
(8). At the end of 2007, the US
National Institutes of Health invested hundreds of millions of
dollars to start a five-year human microbial genome project, mainly
using high-throughput sequencing technology analysis of different
parts of the body's bacterial community structure and the female
vaginal micro-ecological environment is an important part of this
program (9,10). Theoretically, 16S ribosomal (r)RNA
gene sequencing is designed to detect most pathologival bacteria.
Via this method, bacteria that are not detectable by cultivation
and empirical antibiotic treatments are identifiable. In addition,
this method may be utilized to identify novel pathogens that have
not been previously recognized as etiologic agents (11).
The diversity of the microflora in the female
reproductive tract is affected by various drugs, as well as changes
in the environment, and the host's hormone levels and immune system
(12). Numerous studies have
demonstrated that the microbial balance in the reproductive tract
is closely associated with a poor outcome of pregnancy (13–16).
However, due to the complex etiology and pathogenesis of RM, the
association of aberrations in the vaginal flora and RM has remained
to be sufficiently elucidated. In the present study, the diversity
of the vaginal microbial community in patients with unexplained RM
was analyzed in order to determine the involvement of the vaginal
flora in the mechanisms of RM.
Materials and methods
Sample collection
Vaginal secretions of 10 women with RM (case group)
and 10 healthy volunteers (control group) were collected at
Shaoxing Women and Children's Hospital (Shaoxing, China) between
September 2016 and March 2017. The samples were collected in
accordance with the relevant guidelines and all subjects provided
written informed consent for use of their samples in the present
study. The present study was approved by the Ethics Committee of
Shaoxing Women and Children's Hospital (Shaoxing, China). None of
the women were pregnant, while they were of reproductive age (case
group, 20–35 years, median age, 28 years; control group, 26–34
years; median age, 30 years; P<0.05), had a regular menstrual
cycle and a history of sexual activity, and had not taken any
antibiotics or antimycotic drugs in the past 30 days. Women were
asked to refrain from sexual activity for 3 days prior to sampling.
Women were excluded from the study if they had used douches,
vaginal medications or suppositories, feminine sprays, genital
wipes or contraceptive spermicides, or had reported vaginal
discharge in the past 48 h (12).
The secretions were scraped from the vaginal walls of the RM
patients and healthy controls using aseptic swabs. From each
subject, two vaginal swabs were collected. The sample centrifuge
tubes were immediately placed in a prepared ice box or in a foam
box filled with ice packs to maintain a low temperature. The
samples were then transferred to the laboratory and stored at
−80°C.
DNA extraction and polymerase chain
reaction (PCR) amplification
Microbial DNA was extracted from vaginal discharge
using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according
to manufacturer's protocols. The V3-V4 region of the bacterial 16S
rRNA gene was amplified by PCR (ABI GeneAmp® 9700). The
thermocycling protocol was as follows: 95°C for 2 min, followed by
25 cycles at 95°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec
and a final extension at 72°C for 5 min. The primers used were 341F
5′-barcode-CCTACGGGNGGCWGCAG-3′ and 805R
5′-barcode-GACTACHVGGGTATCTAATCC-3′, where the barcode is an
eight-base sequence unique to each sample. PCR was performed in
triplicate in 20 µl mixtures containing 4 µl 5X Fast Pfu Buffer, 2
µl 2.5 mM deoxynucleoside triphosphates, 0.8 µl of each primer (5
µM), 0.4 µl FastPfu Polymerase and 10 ng template DNA (17). All samples were subjected to the same
conditions and each sample was assessed in duplicate. The PCR
products of the same sample were mixed and examined by 2% agarose
gel electrophoresis. The PCR products were recovered using
AxyPrepDNA Gel Recovery Kit (Axygen; Corning, Inc., Corning, NY,
USA) Tris-HCl elution (18).
Illumina MiSeq sequencing
The PCR products were quantified using the
QuantiFluor™-ST Blue Fluorescence Quantitative System (Promega
Corp., Madison, WI, USA) after obtaining the initial quantitative
results of the electrophoresis, followed by mixing the
corresponding proportions according to the sequencing requirements
of each sample. Amplicons were extracted from 2% agarose gels and
purified using the AxyPrep DNA Gel Extraction kit (Axygen; Corning,
Inc.) according to the manufacturer's instructions and quantified
using QuantiFluor™-ST (Promega Corp.). Purified amplicons were
pooled in equimolar amounts and paired-end sequenced (2×250) on an
Illumina MiSeq platform according to the standard protocols.
Processing of sequencing data
Sequences produced by Miseq sequencing contain
barcode sequences, and the primers and linker sequences are added
at the time of sequencing. First, the primer sequences were removed
using Cutadapt and the pairs were merged into a sequence according
to the overlap between paired-end (PE) reads by using The PE reAd
merger (19). Next the sample data
were identified and distinguished according to the barcode tag
sequences. Finally, quality control and filtration of each sample
was performed using Prinseq (20) to
obtain data.
Raw fastq files were demultiplexed and
quality-filtered using FLASH (21)
with the following criteria: i) The 3′ end of the sequencing primer
was removed and the Read1 3′ end sequencing link was
TGGAATTCTCGGGTGCCAAGGAACTC; ii) according to the overlap between PE
reads, pairs of reads were merged into a sequence, and the allowed
maximum mismatch ratio of the splicing sequence overlap area was
0.1; iii) data for each sample were divided from the merged data
according to each sample's barcode sequence; iv) the reads were
truncated at any site receiving an average quality score of <20
over a 10-bp sliding window; if the average quality value of the
window was <20, the back-end base from the window was cut. v)
Reads containing N sequences were removed along with the short
sequence in the data, with a length threshold of 200 bp. vi) The
low-complexity sequences were filtered.
Bacterial community
characterization
According to the barcodes, the high-throughput
pyrosequencing reads from 20 vaginal secretion samples were
reassigned to samples. Operational taxonomic units (OTUs) were
clustered with a 97% similarity cutoff through Usearch (version
7.0; http://drive5.com/uparse/) (22), and chimeric sequences were identified
and removed using UCHIME (23).
Alpha diversity analysis was performed using the OTUs that reached
a similarity level of 97%, and the species diversity in a single
sample was analyzed by evaluation of Chao, abundance-based coverage
estimators (ACE) and Simpon parameters using Mothur (24). A Venn diagram was generated to
illustrate the shared and unique OTUs among the groups, based on
the occurrence of OTUs in a group of samples regardless of their
relative abundance, and this was analyzed using the Venn Diagram
package of R language tools (25).
Beta diversity was analyzed to investigate the
similarity of the bacterial community structure among groups using
Weighted Unifrac distances and visualized analysis via principal
component analysis (PCoA). Circos, a sample and species association
map, is a visual circle diagram that describes the correspondence
between sample and species. The functional profiles of microbial
communities were predicted by using PICRUSt (26). The 10,506 OTUs were normalized to the
16S rRNA copy number and their metagenomic contributions were
predicted according to the Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathways. Based on the PICRUSt functional secondary
classification results, the differences in abundance between
samples were compared.
Statistical analysis
All statistical analyses were performed using R
packages (V.3.2) (25). The
screening criterion for analyzing differences in bacterial
abundance between samples was P≤0.05. Fisher's exact test was used
for comparison between samples and Welch's t-test was used for
comparison between groups (27).
Finally, the P-value was verified by using the false discovery rate
to obtain the q-value.
Results
Alpha diversity analysis between the
case and control groups
To characterize the vaginal microbiota and potential
variations associated with RM, the vaginal secretions from 10 cases
of RM were collected, and those from 10 healthy women were used as
a control group. In total, 745,971 usable sequences from 20 samples
were obtained. A total of 10,506 OTUs were delineated at a 97%
similarity level. On average, >90% of reads in a sample were
retained. At a 97% sequence similarity level, Good's coverage
values for all sampled bacterial communities ranged from 0.989 to
0.995 when estimated from all reads (Table I).
 | Table I.Summary of pyrosequencing data. |
Table I.
Summary of pyrosequencing data.
| Sample_ID | Sequence_no. | OTUs | Filtered OTUs | Shannon_index | ACE_index | Chao1_index | Coverage | Simpon |
|---|
| RM_control1 | 34948 | 447 | 37322 | 1.2204 | 641.11437 | 580.913043 | 0.994964 | 0.56009 |
| RM_control2 | 39002 | 463 | 41410 | 0.421023 | 1672.661269 | 971.683544 | 0.992718 | 0.905217 |
| RM_control3 | 36163 | 551 | 38664 | 0.883533 | 885.603703 | 787.014925 | 0.993032 | 0.717632 |
| RM_control4 | 38540 | 336 | 39876 | 0.384966 | 448.529403 | 407.90625 | 0.996938 | 0.919171 |
| RM_control5 | 33366 | 637 | 35693 | 1.586938 | 874.401789 | 812.506329 | 0.992927 | 0.478691 |
| RM_control6 | 38787 | 377 | 39998 | 1.193842 | 691.127976 | 618.325 | 0.994921 | 0.460157 |
| RM_control7 | 33174 | 421 | 34615 | 1.90784 | 639.519629 | 559.125 | 0.994876 | 0.282237 |
| RM_control8 | 38476 | 604 | 40815 | 0.605582 | 1702.873689 | 1098.161017 | 0.991111 | 0.872647 |
| RM_control9 | 38193 | 418 | 40387 | 0.349364 | 1476.004523 | 827.296296 | 0.993245 | 0.927695 |
| RM_control10 | 39055 | 556 | 41136 | 0.430429 | 1525.391993 | 1033.097087 | 0.99196 | 0.913746 |
| RM_case1 | 38861 | 530 | 40899 | 0.47373 | 1411.786593 | 889.486486 | 0.992718 | 0.901358 |
| RM_case2 | 37973 | 495 | 39464 | 0.969683 | 688.77094 | 613.106383 | 0.995181 | 0.737364 |
| RM_case3 | 32766 | 659 | 34398 | 2.121407 | 1435.948065 | 1057.649635 | 0.989898 | 0.263954 |
| RM_case4 | 38107 | 633 | 40429 | 0.559294 | 1226.25393 | 1100.12782 | 0.990737 | 0.882253 |
| RM_case5 | 36180 | 553 | 37780 | 0.964259 | 1496.239405 | 983 | 0.99168 | 0.62994 |
| RM_case6 | 39523 | 692 | 42178 | 0.492288 | 1817.054442 | 1243.338129 | 0.990082 | 0.904096 |
| RM_case7 | 34778 | 478 | 36739 | 1.20537 | 766.093643 | 675.280992 | 0.993703 | 0.446528 |
| RM_case8 | 39885 | 510 | 41821 | 0.325184 | 1492.01625 | 904.345794 | 0.992704 | 0.94037 |
| RM_case9 | 39854 | 537 | 41768 | 0.576914 | 1407.226829 | 936.794393 | 0.992648 | 0.866492 |
| RM_case10 | 38340 | 609 | 41200 | 0.486695 | 1252.493401 | 1152.128205 | 0.990689 | 0.900224 |
Alpha diversity was applied to analyze the
complexity of the bacterial diversity of the samples. In Fig. 1, a boxplot of richness estimators was
generated using the QIIME toolkit, providing a clear visualization
of the diversities in the different sample groups. No significant
difference in Simpon, ACE and Chao diversity was identified between
the control and case groups in the box plots (P>0.05; Fig. 1). Community richness comparison
indicated that the case group had a significantly increased number
of observed and estimated OTUs compared with those in the control
group (P=0.037). As displayed in the Venn diagram, the groups had
1,924 OTUs in common, and the proportion of unique OTUs among the
total OTUs was 43.8 and 41.9% in the case and control group,
respectively. However, the case group had 63 more OTUs than the
control group (Fig. 2). This result
demonstrated a significantly higher diversity in the case group
compared with that in the control group. As presented in Fig. 3, the Simpon rarefaction curves for
all samples were saturated, which indicated that the 16S rRNA gene
sequence was highly abundant in the database, and that the present
analysis had an adequate depth to retrieve most of the information
on microbial diversity.
Characteristics of beta diversity
analysis between the case and control groups
In the PCoA plot (Fig.
4), each dot represents one sample. PCoA based on 10,506 OTUs
(grouped at a sequence identity of 97%) revealed a slight
separation of the two different groups on the first two PCo scores,
PCo1 and PCo2, which accounted for 36.8 and 29.9% of the total
variations, respectively. This indicated that the development of RM
may contribute to microbiota imbalance. Furthermore, microbiota
disturbances may further promote the development of RM (Fig. 4).
Comparison of bacterial communities in
the case and control groups at the phylum level
When comparing the alpha diversity between the
groups, no significant differences in diversity were observed
except for a higher richness index in the case group compared with
that in the control group. However, the PCoA plot based on Weighted
Unifrac data displayed distinct bacterial community structures in
the case and the control group. To investigate the specific changes
of the microbiota in the samples from the case group, the relative
abundance of taxa in the different groups was assessed. Fig. 5 presents taxonomic distributions of
the predominant bacteria (relative abundance, >1% of the total
sequences) at the phylum level. The bacterial flora analysis
indicated that Firmicutes was the most predominant phylum,
accounting for 79.57 and 73.12% of the microbiota in case and
control group, respectively. The second most dominant phylum was
Actinobacteria (18.86% in the case group and 22.95% in the
control group). Changes at the phylum level were mainly in the
three most common types of bacteria (Firmicutes,
Bacteroidetes and Actinobacteria). Compared with that in
the control group, the relative abundance of Firmicutes in
the case group was increased by 5.72%, while the
Actinobacteria and the Bacteroidetes were reduced by
3.08 and 3.07%, respectively.
Comparison of bacterial communities
between case and control groups at the genus level
The bacterial diversity and relative abundance in
all samples at the genus level are presented in Fig. 6. Fig.
6A displays the composition of the vaginal flora of the
patients with RM and healthy women. In the stacked bar chart, each
bar represents the average relative abundance of each bacterial
taxon. The top 50 taxa with high relative abundance are
illustrated. All core bacteria in the vagina, including
Lactobacillus, Gardnerella, Atopobium and Prevotella,
were identified in the present study. Of all the detected genera,
Lactobacillus of the phylum Firmicutes were the most
abundant ones. Circos was then used to identify bacterial taxa that
were significantly different between the groups. A Circos
presentation of bacterial taxa that are differentially represented
between the different groups is provided in Fig. 6B. In the case group, 3 bacterial taxa
were significantly more abundant (Atopobium, Prevotella and
Streptococcus), while only 2 taxa were overrepresented in
the control group (Lactobacillus and
Gardnerella).
Functional annotation
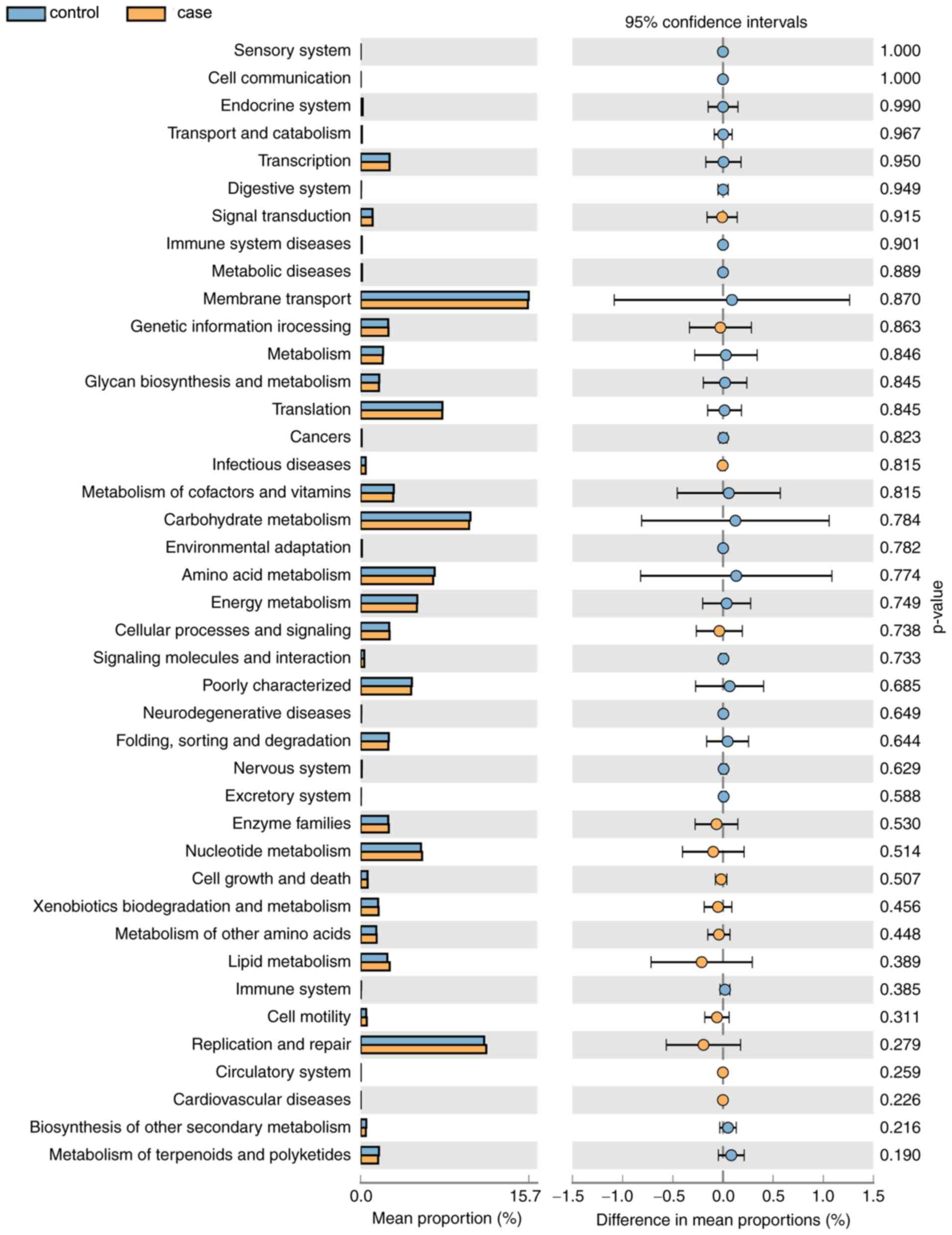
As displayed in Fig.
7, a total of 14 KEGG pathways (e.g. ‘Signal Transduction’ and
‘Cell Motility’) were more abundant in the case group and 27
pathways (e.g. ‘Metabolism of Cofactors and Vitamins’ and
‘Carbohydrate Metabolism’) were more abundant in the control group.
However, the differences in the mean proportion of the abundance of
the functional pathways between the groups were not significant
(P>0.05). In the future, differences in the functional pathways
specific for the vaginal microbial flora of the two groups will be
further experimentally verified.
Discussion
Previous studies on vaginal bacterial communities
suggested that the human vaginal microbiota has a key role in
preventing a number of urogenital diseases, including bacterial
vaginosis, yeast infections, sexually transmitted infections and
urinary tract infections (12,28).
However, studies indicating a direct association between RM and
vaginal microbiota are currently lacking. Compared with the
intestinal flora, the vaginal flora is less diverse, and women of
childbearing age may have ~40 species of bacteria and facultative
anaerobic bacteria, including Lactobacillus, Bacteroides,
Coccidioides, Corynebacterium, Escherichia coli, Velveti and
Gardneria (29). A previous
study sampled and analyzed 396 women from four ethnic groups and
identified that the common constituents of the vaginal flora may be
roughly divided into five categories, of which four are able to
produce a large quantity of lactic acid to generate the acidic
environment in the vagina (30).
Bacterial vaginosis is characterized by a complete loss of
lactobacilli and a concomitant increase in Gram-variable and
Gram-negative rods, with Gardnerella vaginalis, as well as
Bacteroidetes, Prevotella and Mobiluncus species
being primary among them. The presence of an abnormal vaginal
microbiota in early pregnancy is a recognized risk factor for
preterm delivery and low birth weight (31).
In the present study, the vaginal microbiome of the
RM group exhibited an increased richness in species as well as a
significant shift in the overall microbial diversity. A
statistically significant increase in richness was observed in the
RM group (P=0.037). These results suggested the presence of
disorders in the profile of the vaginal microbiome in patients with
RM and miscarriage may be either a cause or an effect of the
altered composition of the vaginal microbiome. The PCo coefficients
obtained in the PCoA analysis of the present study indicated that
RM may be the most important factor contributing to changes in the
vaginal bacterial composition. It was suggested that RM may be
associated with imbalances in the microbiota. Local microbiota
disturbances may further promote the development of RM (32).
When comparing the alpha diversity between samples,
significant differences in diversity were observed in richness
indices. Furthermore, PCoA indicated distinct bacterial community
structures between the two groups. To further clarify the specific
differences in the intestinal microflora between the case and
control groups, taxonomic analysis at the phylum and genus level,
as well as beta diversity analysis were performed. At the phylum
level, Firmicutes was the most predominant phylum, followed
by Actinobacteria and then Bacteroidetes.
Furthermore, at the genus level, Lactobacillus was the most
dominant genus. Statistically significant differences in five
genera were observed. In the case group, three bacterial taxa were
significantly more abundant (Atopobium, Prevotella and
Streptococcus), while only two taxa were overrepresented in
the control group (Lactobacillus and Gardnerella).
Kuon et al (33) reported
that RM patients with elevated peripheral and uterine natural
killer cells suffer more frequently from colonization by
Gardnerella vaginalis and gram-negative anaerobes. McDonald
et al (34) demonstrated that
group B streptococcus is a key pathogen in intrauterine infection
and a frequent cause of spontaneous midgestation abortions.
Lactobacillus is one of the diverse and phylogenetically
heterogeneous orders of lactic acid-producing bacteria that
includes the genus Lactobacillus, as well as the genera
Facklamia, Granulicatella, Leuconostoc, Pediococcus
and Streptococcus (35). The
healthy human vagina is dominated by a variety of
Lactobacillus species, which have an essential role in
protecting women from genital infection. A deficiency in
Lactobacilli may disturb the microbial balance in the
vagina, frequently resulting in the syndrome of bacterial
vaginosis, which is associated with a quantitative and qualitative
shift from normally occurring Lactobacilli to a mixed
microflora dominated by anaerobic bacteria (31). In the present study, the three
anaerobic bacteria of Atopobium, Prevotella and
Streptococcus were more abundant in the case group compared
with those in the control group, which was consistent with the
results of previous studies.
In conclusion, the present study defined a
structural imbalance in the vaginal microbiota of patients with RM,
represented by an increased incidence of Atopobium,
Prevotella and Streptococcus and reduction of
Lactobacillus, which was identified by comparing the vaginal
microbiota composition of RM patients with that of healthy
individuals. PCoA analysis suggested that changes in vaginal flora
may be the cause of/associated with RM. The present results point
towards a novel strategy aimed at preventing the development of RM
through restoring the homeostasis of the vaginal microbiome, by
improvement in lifestyle or early intervention with drugs or
probiotics.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81701522); the
National Natural Science Foundation of Zhejiang Province, China
(grant nos. LY15H040001 and LY16H040008); the Science Technology
Department of Zhejiang Province, China (grant no. 2015C33280,
2016C33222, 2016C33223 and 2018C37102); the Medical and Health
Project of Zhejiang Province, China (grant nos. 2014KYA275,
2014KYA276, 2017KY669, 2017KY670, 2017KY672, 2018KY844, 2018KY845,
2018KY846 and 2018KY848); the Health and Family Planning Commission
of Shaoxing, China (grant nos. 2017QN008 and 2017CX011); and the
Science Technology Department of Shaoxing, China (grant no.
2017B70004).
Availability of data and material
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
FZ, TZ and HD conceived and designed the current
study. YM and ZH obtained samples; YH performed the experiments; HP
collected the data; MF and HD analysed and interpreted the data.
All authors approved the final version of the manuscript for
publication.
Ethical approval and consent to
participate
All subjects provided written informed consent for
use of their samples in the present study. The present study was
approved by the Ethics Committee of Shaoxing Women and Children's
Hospital (Shaoxing, China) and was performed in compliance with the
Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sugiura-Ogasawara M, Ozaki Y and Suzumori
N: Management of recurrent miscarriage. J Obstet Gynaecol Res.
40:1174–1179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dempsey MA, Flood K, Burke N, Murray A,
Cotter B, Mullers S, Dicker P, Fletcher P, Geary M, Kenny D and
Malone FD: Platelet function in patients with a history of
unexplained recurrent miscarriage who subsequently miscarry again.
Eur J Obstet Gynecol Reprod Biol. 188:61–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Christiansen OB, Larsen EC, Egerup P,
Lunoee L, Egestad L and Nielsen HS: Intravenous immunoglobulin
treatment for secondary recurrent miscarriage: A randomised,
double-blind, placebo-controlled tria. BJOG. 122:500–508. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Obiero JA, Waititu KK, Mulei I, Omar FI,
Jaoko W and Mwethera PG: Baboon vaginal microbial flora. J Med
Primatol. 45:147–155. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Farr A, Kiss H, Hagmann M, Machal S,
Holzer I, Kueronya V, Husslein PW and Petricevic L: Role of
lactobacillus species in the intermediate vaginal flora in early
pregnancy: A retrospective cohort study. PLoS One. 10:e01441812015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Romero R, Chaiworapongsa T, Kuivaniemi H
and Tromp G: Bacterial vaginosis, the inflammatory response and the
risk of preterm birth: A role for genetic epidemiology in the
prevention of preterm birth. Am J Obstet Gynecol. 190:1509–1519.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Romero HD and Andreu DA: Bacterial
vaginosis. Enfermedades Infecciosas Y Microbiol Clin. 34 (Suppl
3):14–18. 2016. View Article : Google Scholar
|
|
8
|
Tseng CH, Lin JT, Ho HJ, Lai ZL, Wang CB,
Tang SL and Wu CY: Gastric microbiota and predicted gene functions
are altered after subtotal gastrectomy in patients with gastric
cancer. Sci Rep. 6:207012016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
NIH HMP Working Group, ; Peterson J,
Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V,
McEwen JE, Wetterstrand KA, et al: The NIH human microbiome
project. Genome Res. 19:2317–2323. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Human Microbiome Jumpstart Reference
Strains Consortium, ; Nelson KE, Weinstock GM, Highlander SK,
Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E,
et al: A catalog of reference genomes from the human microbiome.
Science. 328:994–999. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ling Z, Liu X, Chen X, Zhu H, Nelson KE,
Xia Y, Li L and Xiang C: Diversity of cervicovaginal microbiota
associated with female lower genital tract infections. Microb Ecol.
61:704–714. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ravel J, Gajer P, Abdo Z, Schneider GM,
Koenig SS, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO,
et al: Vaginal microbiome of reproductive-age women. Proc Natl Acad
Sci USA. 108 (Suppl 1):4680–4687. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leitch H and Kiss H: Asymptomatic
bacterial vaginosis and intermediate flora as risk factors for
adverse pregnancy outcome. Best Pract Res Clin Obstet Gynaecol.
21:375–390. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Allsworth JE and Peipert JF: Prevalence of
bacterial vaginosis: 2001–2004 National Health and Nutrition
Examination Survey data. Obstet Gynecol. 109:114–120. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trabert B and Misra DP: Risk factors for
bacterial vaginosis during pregnancy among African American women.
Am J Obstet Gynecol. 197:477.e1–e8. 2007. View Article : Google Scholar
|
|
16
|
Donders GG, Veerecken A, Bosmans E,
Dekersmaecker A, Salembier G and Spitz B: Definition of a type of
abnormal vaginal flora that is distinct from bacterial vaginosis:
Aerobic vaginitis. BJOG. 109:34–43. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Caporaso JG, Lauber CL, Walters WA,
Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N and Knight R:
Global patterns of 16S rRNA diversity at a depth of millions of
sequences per sample. Proc Natl Acad Sci USA. 108 (Suppl
1):4516–4522. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aguirre E, Galiana A, Mira A, Guardiola R,
Sánchez-Guillén L, Garcia-Pachon E, Santibañez M, Royo G and
Rodríguez JC: Analysis of microbiota in stable patients with
chronic obstructive pulmonary disease. APMIS. 123:427–432. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Kobert K, Flouri T and Stamatakis
A: PEAR: A fast and accurate Illumina Paired-End reAd mergeR.
Bioinformatics. 30:614–620. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schmieder R and Edwards R: Quality control
and preprocessing of metagenomic datasets. Bioinformatics.
27:863–864. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Magoč T and Salzberg SL: FLASH: Fast
length adjustment of short reads to improve genome assemblies.
Bioinformatics. 27:2957–2963. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Edgar RC: Search and clustering orders of
magnitude faster than BLAST. Bioinformatics. 26:2460–2461. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Edgar RC, Haas BJ, Clemente JC, Quince C
and Knight R: UCHIME improves sensitivity and speed of chimera
detection. Bioinformatics. 27:2194–2200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schloss PD, Westcott SL, Ryabin T, Hall
JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH,
Robinson CJ, et al: Introducing mothur: Open-source,
platform-independent, community-supported software for describing
and comparing microbial communities. Appl Environ Microbiol.
75:7537–7541. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Snipen L and Liland KH: micropan: An
R-package for microbial pan-genomics. BMC Bioinformatics.
16:792015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Langille MG, Zaneveld J, Caporaso JG,
McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega
Thurber RL, Knight R, et al: Predictive functional profiling of
microbial communities using 16S rRNA marker gene sequences. Nat
Biotechnol. 31:814–821. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Parks DH, Tyson GW, Hugenholtz P and Beiko
RG: STAMP: Statistical analysis of taxonomic and functional
profiles. Bioinformatics. 30:3123–3124. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hernándezrodríguez C, Romerogonzález R,
Albanicampanario M, Figueroa-Damián R, Meraz-Cruz N and
Hernández-Guerrero C: Vaginal microbiota of healthy pregnant
mexican women is constituted by four lactobacillus species and,
several vaginosis-associated bacteria. Infect Dis Obstet Gynecol.
2011:8514852011.PubMed/NCBI
|
|
29
|
Wells CL, Jechorek RP and Maddaus MA: The
translocation of intestinal facultative and anaerobic bacteria in
defined flora mice. Microb Eco Health Dis. 1:227–235. 1988.
|
|
30
|
Mendes-Soares H, Suzuki H, Hickey RJ and
Forney LJ: Comparative functional genomics of Lactobacillus spp.
reveals possible mechanisms for specialization of vaginal
lactobacilli to their environment. J Bacteriol. 196:1458–1470.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Petricevic L, Domig KJ, Nierscher FJ,
Sandhofer MJ, Fidesser M, Krondorfer I, Husslein P, Kneifel W and
Kiss H: Characterisation of the vaginal Lactobacillus microbiota
associated with preterm delivery. Sci Rep. 4:51362014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu Y, Chen J, Zheng J, Hu G, Wang J, Huang
C, Lou L, Wang X and Zeng Y: Mucosal adherent bacterial dysbiosis
in patients with colorectal adenomas. Sci Rep. 6:263372016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kuon RJ, Togawa R, Vomstein K, Weber M,
Goeggl T, Strowitzki T, Markert UR, Zimmermann S, Daniel V, Dalpke
AH and Toth B: Higher prevalence of colonization with gardnerella
vaginalis and gram-negative anaerobes in patients with recurrent
miscarriage and elevated peripheral natural killer cells. J Reprod
Immunol. 120:15–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mcdonald HM and Chambers HM: Intrauterine
infection and spontaneous midgestation abortion: Is the spectrum of
microorganisms similar to that in preterm labor? Infect Dis Obst
Gynecol. 8:220–227. 2000. View Article : Google Scholar
|
|
35
|
Goldstein EJ, Tyrrell KL and Citron DM:
Lactobacillus species: Taxonomic complexity and controversial
susceptibilities. Clin Infect Dis. 60 (Suppl 2):S98–S107. 2015.
View Article : Google Scholar : PubMed/NCBI
|