Introduction
DNA methylation serves an essential role in the
stability and regulation of gene expression during development and
in the maintenance of cellular identity (1). Dynamic changes in DNA methylation are
essential to mammalian development as they contribute to cell
growth, differentiation, and in particular, early embryonic
development (2,3). DNA methylation is associated with
critical biological functions, including genomic imprinting,
inactivation of the X chromosome and the regulation of gene
expression (4–6). DNA methylation is a chemical
modification that typically occurs within a CpG dinucleotide region
in adult somatic cells (7,8). The hypermethylation of promoter CpG
islands affects tumor suppressive mRNAs (9–10).
Methylation of promoter CpG islands present in or near promoter
regions may also disrupt the binding of transcription factors
(11). Epigenetic modifications,
including DNA methylation patterns, represent epigenetic barriers
that limit developmental potential during mammalian development
(12). Dramatic changes in
epigenetic status enable the zygote to erase epigenetic signatures
that are inherited from the gametes and subsequently obtain
developmental totipotency (12).
Recent advances have begun to elucidate how such
dramatic demethylation is activated in the zygote, but a clear
picture of the mechanistic details has not yet emerged. Smallwood
et al (13) performed the
first integrated epigenomic analysis of mammalian oocytes and
identified over 1,000 CpG islands that were methylated in mature
oocytes. They also demonstrated that methylation of these CpG
islands was dependent on DNA methyltransferase 3α and DNA
methyltransferase 3-like, but the methylation changes were not
distinct at the sequence level. Following fertilization,
methylation is comprehensively reprogrammed (9). The maternal genome is demethylated
passively and the paternal genome is activated in the zygote during
the developmental process of the zygote and morula (9,13).
Therefore, the establishment of novel methylation landscapes
begins. These results provide an important insight into the
mechanisms of methylation in germ cells. However, DNA methylome
establishment and maintenance in human sperm, oocytes and various
developmental stages of the preimplantation embryo have not been
described in detail. This is partly due to technical limitations of
genome-wide studies in cells, yet this area deserves further
exploration (14).
Primordial germ cell differentiation to produce
mature gametes, and genome transformation following fertilization
are the two important phases of the mammalian life cycle. In
previous studies (15,16), quantitative analysis was performed on
whole genome methylation sites in human and mammalian gametes and
early embryos, which demonstrated the generation of mature gametes
in human and mammal fertilized zygotes and DNA methylation and
exhibited dynamic changes during preimplantation embryo
development. These dynamic patterns of methylation provide an
important theoretical basis for understanding gene expression and
regulation of the early human embryo, as well as the inhibitory
effects of transposons. However, the majority of previous studies
used the reduced representation bisulfite sequencing (RRBS) method.
Although RRBS can accurately distinguish 5-mC and 5-hmC (10), the coverage rate is low (10% CpG
island of the methylation sites) (17). Guo et al (17) used the method of whole genome
bisulphite sequencing (WGBS) to detect methylation site changes of
the inner cell mass (ICM) and embryo. However, they did not
elucidate genome-wide methylation site changes of gametes and the
blastula.
The methylated DNA immunoprecipitation (MeDIP) -Chip
method can cover almost all promoters and CpG islands (18). In the present study, MeDIP-Chip was
performed to analyze dynamic changes in whole genome CpG island
methylation at various developmental stages of human sperm, oocytes
and preimplantation embryos. The current study could improve
understanding of the CpG island and promoter methylation pattern
during early embryonic development.
Materials and methods
Patient information
A total of 43 healthy couples and volunteers
(average age, 28.93±4.15 years; male: female, 24:19) were recruited
at the Third Affiliated Hospital of Guangzhou Medical University
(Guangzhou, China) between September 2010 and November 2016. The
inclusion criteria were as follows: i) normal density, vigor and
deformity rate in male semen and ii) normal female ovarian
function, according to the World Health Organisation criteria for
human semen (5th edition) testing standards (19). The exclusion criteria were as
follows: i) age >40 years, ii) polycystic ovary syndrome,
premature ovarian failure, endometrial polyps or endometriosis, and
iii) smoker or alcoholic.
Grouping information
Samples were divided into eight groups based on
developmental stage: 1, Sperm group (n=30); 2, MII oocyte group
(n=25); 3, two-pronuclei (2PN) period zygote group (n=25); 4,
4-cell stage embryo group (n=5); 5, 8-cell stage embryo group
(n=4); 6, morula embryo group (n=6); 7, blastular ICM group (n=5);
and 8, blastular trophoblastic cells (TE) group (n=5).
Oocyte collection and DNA
extraction
Oocytes were collected from patients and volunteers
at The Third Affiliated Hospital of Guangzhou Medical University
between January 2010 and December 2015.
Under a stereomicroscope, individual eggs (~140 µm
in diameter) were collected and placed in Tyrode's solution (SAGE;
Cooper Surgical, Trumball, CT, USA). Following the disappearance of
the zona pellucida observed under a stereomicroscope, the oocytes
(~0.1 ml/drop) were placed in a buffer containing 2 µl lysis buffer
A (10 mM Tris-HCl, 1 mM EDTA, 10% SDS and 1% proteinase K; pH 8.0).
The cells were placed in a water bath at 37°C for ~1 h to fully
lyse the cells. The cells were centrifuged at 11,180 × g for 2 min
at room temperature for 2 min and stored at −80°C until use. A
total of 2 µl of the last wash liquid was taken for the polymerase
chain reaction (PCR) blank control.
Sperm collection and DNA
extraction
The sperm of patients who had accepted in
vitro fertilization (IVF) treatment at Guangzhou Medical Third
Affiliated Hospital between January 2010 and December 2015 were
collected. The patient age ranged between 22 and 40 years. These
couples' infertility was not caused by any male factor in semen.
Density gradient centrifugation at 400 × g for 10 min at −4°C and
upstream separation were applied for collection of sperm. The
differential lysis method (20) was
used, and DNA was extracted using the Genomic DNA Purification Kit
(Promega Corporation, Madison, WI, USA) according to the kit
protocol, in combination with reagents.
2PN and 4-cell embryo collection and
DNA extraction
Intracytoplasmic sperm injection (ICSI)
fertilization was performed using oocytes and spermatozoa from the
aforementioned sources, and the mature egg was selected. The egg
droplet was moved into the field of view. Securing the egg with the
holding pipette, the oocyte was transferred to the focal plane
under microinjection needle. The polar body was located at the 6–7
or 11–12 o'clock position, so that the injection of the
quasi-oocyte was at the 3 o'clock position.
The sperm were pushed to the tip of the injection
needle. The injection needle was inserted into the oocyte 3 o'clock
position and through the zona pellucida, until the sperm was pushed
within the cytoplasm of the middle of the egg. Once the eggs had
been injected, they were repeatedly rinsed in a pre-prepared G1.5
Plus Petri dish and placed in incubators containing 5%
CO2, 5% O2 and 90% N2 at 37°C and
cultured in G1.5 Plus droplets separately.
For IVF, GIVF-Plus medium (Vitrolife AB, Göteborg,
Sweden) that had been equilibrated overnight was used to prepare
fertilized droplets according to the standard of 0.1 ml per
microtube droplet per 3 eggs. These fertilized droplets were stored
in an incubator (37°C, 6% CO2) with a covering of
mineral oil. A moderate amount of sperm was added to a prepared
fertilization dish under a microscope. The concentration was
adjusted to 1.0×106/ml. These sperm droplets were stored
in an incubator (37°C, 6% CO2) until fertilization.
Following 3–4 h of pre-incubation, eggs were
transferred to the fertilized dish. Two or three eggs were added to
one semen drop and this fertilized drop was returned to the embryo
box for overnight cultivation.
For 2PN collection, prokaryotic cells were observed
at 16–20 h following ICSI or IVF under the inverted microscope and
fertilization information was recorded. Two circular structures
with nucleolar precursors in the cytoplasm indicated the male and
female pronuclei (2PN). Two polar bodies could be observed in the
perivitelline space when normal fertilization occurred. DNA was
extracted from 12 prokaryotic and ICSI-derived prokaryotes from
2PN-derived IVF using the DNA extraction method as previously
described for oocytes.
Two prokaryotic embryos were maintained in culture
to D2 at 16–20 h following ICSI or IVF. These embryos continued to
culture until embryos had 4 cells, uniform blastomere size and a
low fragmentation rate (<5%) was met. A total of 5 embryos were
subsequently used in present study. The DNA extraction method was
identical to that used for oocytes.
D3 embryo collection and DNA
extraction
Following 2 years of successful transplants, donated
embryos were stored via the vitrified cryopreservation method. Once
thawed, there were 7–9 available embryos, which had uniform size
and a low cell debris rate (<20%). Five of these embryos were
used in the present study. The DNA extraction method was identical
to that used for oocytes.
Morula collection and DNA
extraction
The thawed D3 embryos were maintained in culture in
incubators containing 5% CO2, 5% O2 and 90%
N2 at 37°C. Following overnight culturing, six morula
embryos, which were completely fused and with <20%
fragmentation, were used in the present study. The DNA extraction
method was identical to that used for oocytes.
Blastocyst ICM and TE collection and
DNA extraction
The aforementioned thawed D3 embryos were cultured
to D5 embryos, and high-quality blastocysts were screened.
Screening of D5 high-quality blastocysts was based on Garden's
grading standards: Blastocysts were grade A or B. Separation of the
ICM and TE was performed via mechanical methods; a capillary
pipette was used to segregate the ICM and TE under a
stereomicroscope. The isolated ICM and TE were washed several times
in phosphate-buffered saline and then DNA was extracted using the
aforementioned method for oocyte DNA extraction.
MeDIP-Chip
Genomic DNA was extracted using the
Wizard® Genomic DNA Purification kit (Promega
Corporation), according to the manufacturer's protocol and
sonicated to random fragments of 200–1,000 bp. Immunoprecipitation
of methylated DNA was performed using Biomag magnetic beads
(Polysciences, Inc., Warrington, PA, USA) coupled with a mouse
monoclonal 5-methylcytosine antibody (1:100; cat no. C15200081-100;
Diagenode, Seraing (Ougrée), Belgium) for 2 h at 4°C. DNA was
purified by phenol/chloroform extraction and ethanol precipitation.
Immunoprecipitated and input DNA were labeled with Cy3- and
Cy5-labeled random 9-mers, respectively, and hybridized to a
NimbleGen Human DNA Methylation 3×720K CpG Island Plus RefSeq
Promoter Microarray (Roche Diagnostics, Basel, Switzerland). This
is a multiplex slide with three identical arrays per slide, and
each array includes 27,728 CpG island regions (from approximately
−2,440 to +610 bp from the transcription start sites) fully covered
by ~720,000 probes. Scanning was performed with the Axon GenePix
4000B microarray scanner (Molecular Devices, LLC, Sunnyvale, CA,
USA).
Statistical analysis
Raw data were extracted as paired files by
NimbleScan software V2.5 (Roche Diagnostics). Median-centering,
quantile normalization and linear smoothing were performed by
Bioconductor packages Ringo (www.bioconductor.org/packages/release/bioc/html/Ringo.html),
limma (http://www.bioconductor.org/packages/release/bioc/html/limma.html)
and MEDME (www.bioconductor.org/packages/release/bioc/html/MEDME.html).
Following normalization, performing median-centering and quantile
normalization using Bioconductor packages Ringo and limma generated
normalized log2-ratio data. A modified ACME algorithm (21) was employed where a fixed-length
window was slid along the length of each chromosome, testing at
each probe using a one-sided Kolmogorov-Smirnov test whether the
surrounding window was enriched for high-intensity probes relative
to the rest of the array. Each probe had a corresponding P-value
score (-log10) and a threshold was set to select regions that were
enriched in the test sample. From the normalized log2-ratio data, a
sliding-window peak-finding algorithm provided by NimbleScan v2.5
(Roche Diagnostics) was applied to identify the enriched peaks with
specified parameters (sliding window width, 750 bp; mini probes per
peak, 2; P-value minimum cut-off=2; maximum spacing between nearby
probes within peak, 500 bp). The identified peaks were mapped to
genomic features: Transcripts and CpG islands.
Results
Dynamic changes in whole-genome CpG
island methylation of human preimplantation embryos
CpG island-associated peak M-value was calculated as
a semi-quantitative indicator of the level of CpG island
methylation in human sperm, oocytes and various developmental
stages of preimplantation embryo. The peak M-value of sperm was
highest (n=15,604), followed by oocytes (n=6,062). The peak M-value
of zygotes decreased from 2PN stage (n=3,744) and reached its
minimum level at the 4-cell stage (n=2,826). This peak M-value
began to rise from 8-cell stage (n=3,073) to morula stage (n=5,374)
and to ICM stage (n=5,706) and TE stage (n=8,376; Fig. 1). The peak M-value of the ICM stage
was similar to that of oocytes. The peak M-value of TE fell between
those of oocytes and sperm.
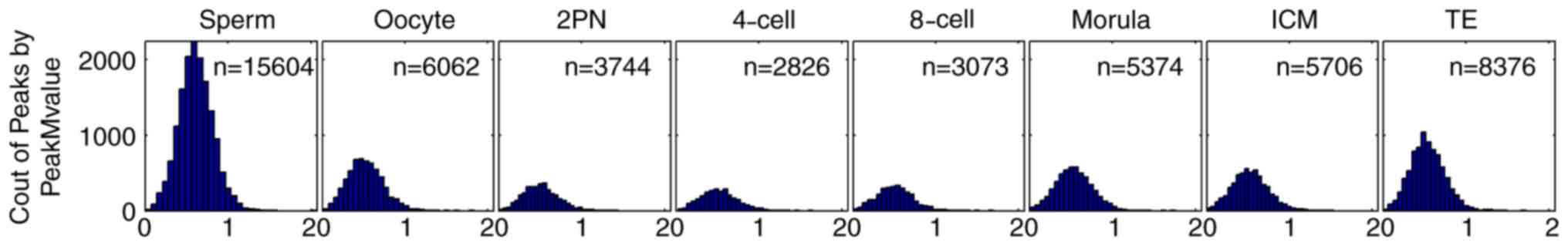 | Figure 1.CpG island-associated peak M-value in
sperm (n=15,604), oocyte (n=6,062), 2PN stage (n=3,744), 4-cell
stage (n=2,826), 8-cell stage (n=3,073), morula stage (n=5,374),
ICM (n=5,706) and TE (n=8,376). Horizontal axis: CpG-island
associated peak M-value. The peak M-value of sperm was highest
(n=15,604), followed by oocytes (n=6,062). The peak M-value of
zygotes decreased from 2PN stage (n=3,744) and reached its minimum
level at the 4-cell stage (n=2,826). This peak M-value began to
rise from 8-cell stage (n=3,073) to morula stage (n=5,374) and to
ICM stage (n=5,706) and TE stage (n=8,376). The peak M-value of the
ICM stage was similar to that of oocytes. The peak M-value of TE
fell between those of oocytes and sperm. ICM, inner cell mass; TE,
trophoblast cells; 2PN, two pronuclei. |
Dynamic changes in the whole-genome CpG island
methylation pattern of human preimplantation embryos were
characterized by low (peak M-value ≤0.4) and high (peak M-value
≥0.7) CpG island methylation regions. The proportion of high CpG
island methylation regions (peak M-value ≥0.7) in 2PN stage was
lowest. After the 2PN stage, CpG island methylation tended to
increase. The proportion of high CpG island methylation regions in
the blastular ICM stage was similar to those in the TE stage.
However, the results for the proportion of low CpG island
methylation regions (peak M-value ≤0.4) were reversed. The
proportion of low CpG island methylation regions (peak M-value
≤0.4) in 2PN stage was highest. Changes in the proportion of middle
CpG island methylation regions were not evident (Table I and Fig.
2).
 | Table I.Proportion of low, middle and high
CpG island-associated peak M-values. |
Table I.
Proportion of low, middle and high
CpG island-associated peak M-values.
|
| Stage (%) |
|---|
|
|
|
|---|
| Peak M-value | Sperm | Oocyte | 2PN | 4-cell | 8-cell | Morula | ICM | TE |
|---|
| Low | 13.90 | 28.84 | 32.05 | 30.33 | 32.18 | 26.57 | 28.58 | 25.76 |
| Middle | 34.77 | 36.89 | 37.39 | 36.41 | 36.45 | 36.30 | 35.96 | 38.44 |
| High | 51.33 | 34.28 | 30.56 | 33.26 | 31.37 | 37.12 | 35.45 | 35.79 |
Variations in methylation level of
whole genome CpG islands of preimplantation embryos
In the first stage from fertilization to 2PN, the
level of CpG island methylation declined sharply. In the second
stage from morula to blastular ICM, methylation rapidly increased
again. The third stage was the methylation reestablishment process
of TE (Table II and Fig. 3).
| Table II.Gene CpG island methylation changes
from gametes to various developmental stages of the preimplantation
embryo. |
Table II.
Gene CpG island methylation changes
from gametes to various developmental stages of the preimplantation
embryo.
| Trend | Sp to 2PN | Oo to 2PN | 2PN to 4-cell | 4-cell to
8-cell | 8-cell to
morula | Morula to ICM | ICM to TE |
|---|
| Stable (n) | 14,397 | 6,277 | 4,336 | 3,965 | 6,401 | 6,096 | 8,086 |
| Changing (n) |
2,102 | 1,656 | 1,070 |
931 | 923 | 2,330 | 2,776 |
Dynamic changes in CpG island
methylation patterns in intragenic, intergenic and promoter
regions
The CpG island methylation levels of zygotes
decreased from the 2PN stage and reached a minimum level at the
4-cell stage. The methylation level began to rise from the 8-cell
stage to the ICM stage, reaching a similar level to the oocyte.
These dynamic changes were derived from methylation in the promoter
region. The proportion of sperm methylation signal in the promoter
region was 73.7%, and that in the oocyte was 60.8%, 2PN was 57.9%,
4-cell stage was 52.2%, 8-cell stage was 50.3%, morula stage was
68.8%, ICM was 66.6% and TE was 66.8%. Methylation fluctuation in
the intergenic region was less obvious than those in promoter
region. However, significant fluctuation of dynamic methylation
changes in intragenic regions were not observed (Fig. 4).
 | Figure 4.CpG island-associated PeakScore in
intragenic, intergenic and promoter regions. (A) Sperm, (B) oocyte,
(C) 2PN stage, (D) 4-cell stage, (E) 8-cell stage, (F) morula
stage, (G) ICM and (H) TE. The proportion of sperm methylation
signal in the promoter region was 73.7%, and that in the oocyte was
60.8%, 2PN was 57.9%, 4-cell stage was 52.2%, 8-cell stage was
50.3%, morula stage was 68.8%, ICM was 66.6% and TE was 66.8%.
Methylation in the intergenic region took second place. However,
dynamic methylation changes in intragenic regions were not
observed. ICM, inner cell mass; TE, trophoblast cells; 2PN, two
pronuclei. |
The mean values of CpG island-associated PeakScore
indicated that dynamic demethylation changes in intragenic,
intergenic and promoter regions were all observed in the
transformation process between sperm, oocyte and zygote (Table III and Fig. 5).
| Table III.CpG island-associated PeakScore in
intragenic, intergenic and promoter regions. |
Table III.
CpG island-associated PeakScore in
intragenic, intergenic and promoter regions.
|
| Stage |
|---|
|
|
|
|---|
| Region | Sperm | Oocyte | 2PN | 4-cell | 8-cell | Morula | ICM | TE |
|---|
| Intergenic, n
(%) | 2,824 (18.1) | 1,542 (25.4) | 1,033 (27.6) | 904 (32.0) | 1,133 (32.6) | 1,133 (21.1) | 1,234 (21.6) | 1,833 (21.9) |
| Intragenic, n
(%) | 1,274 (8.2) | 832 (13.7) | 543 (14.5) | 448 (15.9) | 652 (17.1) | 652 (12.1) | 672 (11.8) | 950 (11.3) |
| Promoter, n
(%) | 11,506 (73.7) | 3,688 (60.8) | 2,168 (57.9) | 1,474 (52.2) | 3,589 (50.3) | 3,589 (68.8) | 3,800 (66.6) | 5,593 (66.8) |
Fluctuation at various stages of the preimplantation
embryo was primarily evident in the promoter region. In promoter
regions, the methylation level reached a minimum value in the 2PN
stage, and subsequently began to rise. The methylation level of ICM
and TE in the promoter region fell between the levels observed in
oocytes and sperm. The CpG methylation fluctuation pattern in
intragenic regions was similar to those in intergenic regions. In
intragenic and intergenic regions, the methylation level decreased
from 2PN stage and reached a minimum value at the 4-cell stage, and
subsequently began to rise. The methylation level of ICM and TE in
these regions was similar to that in oocytes (Fig. 5).
Discussion
DNA methylation, an enzymatic modification at the
5′position of a cytosine pyrimidine ring, is of great importance in
cellular processes, including genome development and regulation
(22). This modification may recruit
methyl-CpG binding proteins to act as a ‘silencing’ epigenetic mark
(23). Changes in DNA methylation
patterns are important in investigating the roles of epigenetics in
the pathogenesis of diseases, and the patterns are subject to
complex regulation during reprogramming (24,25).
Advances in the field of DNA methylation have been made due to the
development of sequencing technology. The majority of previous
studies have either focused on global DNA methylation, low
resolution or candidate gene DNA methylation changes using
sequencing methods, such as bisulfite pyrosequencing (26) and RRBS (27). RRBS is one of several sequencing
methods applied to profile DNA methylation (27). Although RRBS can accurately
distinguish 5-mC and 5-hmC, it only covers 10% of all CpG sites,
which may leave CpG-sparse regions unexplored in the human genome
(15). To improve coverage rate and
obtain absolute quantification of DNA methylation, Guo et al
(15) performed WGBS on the
blastular ICM and post-implantation embryos. However, this
sequencing method requires a higher DNA input. The study did not
elucidate genome-wide methylation site changes of gametes and
blastular embryos, which are difficult to collect. Post-Bisulfite
Adaptor Tagging (PBAT) was also applied to profile DNA methylation
(28). The PBAT method could
generate a substantial number of unamplified reads from as little
as subnanogram quantities of DNA (29). However, site preferences in the
random priming steps would lead to ‘pile-ups’ of reads (29). Differential priming between
methylated and unmethylated alleles may lead to inaccurate
estimation of methylation level (29).
MeDIP sequencing is a popular 5mC capture-based
method, which can detect genome-wide DNA methylation levels rapidly
and cost-efficiently at a resolution of 100–500 bp (30). This sequencing method requires
low-input DNA and has broad application prospects in the study of
DNA methylation (24). In the
present study, MeDIP-Chip was performed to investigate dynamic
changes in whole genome CpG island methylation in human sperm,
oocytes and various developmental stages of the preimplantation
embryo.
The majority of array-based studies are performed
based on immunoprecipitation of methylated DNA, coupled with
hybridization to MeDIP-chip (27,31).
MeDIP-Chip may be a sufficient tool to detect differentially
methylated regions at the level of several hundred base pairs
rather than at the level of single cytosines (32). Among several DNA methylation analysis
methods, MeDIP-chip was previously reported to be a suitable method
to detect methylated DNA information when taking into account cost,
ease of implementation and sensitivity (33,34).
This technology performs well in detecting DNA methylation
information at probe-level resolution, with a low genome-wide
combined false-positive and false-negative rate of approximately
0.21 (32). However, detection of
DNA methylation information can be susceptible to strong signal
distortions, which result from dye bias and the CG content of
effectively unmethylated genomic regions (32).
The current results demonstrated that dynamic
changes in the pattern of CpG island methylation were present in
human sperm, oocytes and various developmental stages of the
preimplantation embryo. The level of CpG island methylation in
human sperm was highest among all the experimental groups.
Demethylation in the zygote began from the pronucleus stage and
methylation reached a minimum level at the 4-cell stage following
fertilization. Methylation was then increased until it was
reestablished at the blastular ICM stage. The level of CpG island
methylation in TE was between that of oocytes and sperm. This
pattern of methylation change was consistent with previous studies:
Methylation in human or mammalian gametes and early embryos was
previously identified to change over different developmental
stages, but there are differences in the pattern of changes between
species (15,35,36). In
genome-scale maps of DNA methylation in gametes and over the
preimplantation timeline, demethylation of the preimplantation
embryo in mice began from the pronuclear stage. A gradual decrease
in methylation was observed from the zygote through the pronuclear
stage and into the blastular ICM, where methylation levels reached
a minimum (35).
Methylation patterns are similar between mouse and
human embryos. In genome-scale maps of DNA methylation in the human
preimplantation embryo, methylation levels of the blastular ICM
exhibited the lowest values (15).
However, genome-wide demethylation in mouse embryos predominantly
occurs at the 1-cell stage, while demethylation in the human embryo
occurs from fertilization to the 2-cell stage (15). In the present study, the lowest
methylation level occurred at the 4-cell stage. This result
indicated that the change in CpG island methylation was primarily
due to CpG island methylation fluctuation in promoter regions,
suggesting that CpG island methylation of the 4-cell stage
primarily occurs in promoter regions. Methylation levels in
promoter regions have been demonstrated to be associated with the
transcriptional activity of genes (37). Therefore, CpG island methylation at
the 4-cell stage may serve an important role in the conversion of
maternal genes to zygotic genes. Various DNA regions of oocytes are
in a demethylated state (38,39). The
methylation status of oocytes is a powerful predictor of zygotic
methylation level and is thought to dictate the zygotic methylation
landscape (35). Hypomethylated
regions in the oocyte could indicate disparities between the sperm
and early embryo (35).
Sperm contribute to the methylation patterns of the
zygote by altering the methylation level of some specific
retroelement subfamilies (35).
Disparities between sperm and oocytes result in different
expression patterns due to epigenetics. Mammalian sperm present
with a high DNA methylation level (35,36).
However, the DNA methylation levels of mammalian and human oocytes
are lower compared with those in sperm (35,36).
Following fertilization, sperm and oocyte DNA in the zygote
undergoes a series of changes. Erasure of DNA methylation may be an
important mechanism in early embryo development. The results of the
current study demonstrated that the DNA CpG island methylation
pattern in the blastular ICM, the final stage before embryo
implantation, is similar to that in oocytes. Compared with sperm,
the CpG island methylation pattern in oocytes is more
significant.
The current results provide insight into the dynamic
changes in whole genome CpG island methylation in human sperm,
oocytes and various developmental stages of preimplantation embryo.
The results indicated that the level of CpG island methylation in
human sperm was higher compared with oocytes and various
developmental stages of the preimplantation embryo. Demethylation
in the zygote began from the pronucleus stage and methylation
reached a minimum level at the 4-cell stage following
fertilization. Methylation then increased until it was
reestablished at the blastular ICM stage, at which point the
methylation level was similar to that in oocytes. The level of CpG
island methylation in TE was between that of oocytes and sperm. The
global methylation level of the preimplantation embryo reached its
minimum value at the ICM stage. It was noted that CpG methylation
erasure of the preimplantation embryo primarily appeared during the
4-cell stage, and whole genome erasure primarily appeared in the
2-cell stage. It was also demonstrated that dynamic changes in CpG
methylation were derived from methylation of promoter regions. The
current MeDIP-Chip analysis results provide an insight into the
dynamic methylation patterns of whole genome CpG islands and
methylation in human sperm, oocytes and embryos.
In conclusion, the current study suggested that CpG
island methylation changes in human preimplantation embryos were
divided into three stages. In the first stage from fertilization to
2PN, the level of CpG island methylation declined sharply. In the
second stage from the morula to the blastular ICM, methylation
rapidly increased again. In the third stage, methylation was
reestablished in the TE. Dynamic CpG island methylation changes
were primarily derived from methylation in promoter regions;
however further validation experiments are required to examine the
methylation variability in larger cohorts. The current study
therefore provides a basis for further epigenetic studies focused
on early zygote development.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Guangdong Natural Science Youth Fund (grant no 2018A030310167),
Guangdong Provincial Department of Education Featured Innovation
Project (grant no. 2017KTSCX153), Guangzhou Liwan District Science
and Technology Plan Project (grant no. 201704037), the National
Nature Science Foundation of China (grant no. 81502507), the
National Nature Science Foundation of China (grant no. 81871211)
and the National Nature Science Foundation of China (grant no.
81801532).
Availability of data and materials
The microarray data (GSE124358) is freely available
from the GEO datasets of the NCBI website (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE124358).
Analysis of these microarray data for this study will be made
available from the corresponding author on reasonable request.
Authors' contributions
LL and YH contributed to the conception and
coordination of the study. YH and HL designed the study and
prepared the manuscript. YH, HL, HD, WZ and XK analyzed the data
and interpreted the results. YL and XZ collected and processed the
samples. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Reproductive
Study Ethics Committee of the Third Affiliated Hospital of
Guangzhou Medical University (Guangzhou, China). All sperm, embryos
were collected from donor couples that provided written informed
consent. Oocyte samples were collected from donor couples or
volunteers that provided their written informed consent.
Patient consent for publication
All patients or volunteers recruited provided
written informed consent for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stelzer Y, Shivalila CS, Soldner F,
Markoulaki S and Jaenisch R: Tracing dynamic changes of DNA
methylation at single-cell resolution. Cell. 163:218–229. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smith ZD and Meissner A: DNA methylation:
Roles in mammalian development. Nat Rev Genet. 14:204–220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
He W, Kang X, Du H, Song B, Lu Z, Huang Y,
Wang D, Sun X, Yu Y and Fan Y: Defining differentially methylated
regions specific for the acquisition of pluripotency and
maintenance in human pluripotent stem cells via microarray. PLoS
One. 9:e1083502014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jaenisch R and Bird A: Epigenetic
regulation of gene expression: How the genome integrates intrinsic
and environmental signals. Nat Genet (33 Suppl). S245–S254. 2003.
View Article : Google Scholar
|
|
5
|
Dhiman VK, Attwood K, Campbell MJ and
Smiraglia DJ: Hormone stimulation of androgen receptor mediates
dynamic changes in DNA methylation patterns at regulatory elements.
Oncotarget. 6:42575–42589. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ping W, Hu J, Hu G, Song Y, Xia Q, Yao M,
Gong S, Jiang C and Yao H: Genome-wide DNA methylation analysis
reveals that mouse chemical iPSCs have closer epigenetic features
to mESCs than OSKM-integrated iPSCs. Cell Death and Disease.
9:1872018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baylin SB: DNA methylation and gene
silencing in cancer. Nat Clin Pract Oncol 1 (2 Suppl). S4–S11.
2005. View Article : Google Scholar
|
|
8
|
Li HJ, Wan RP, Tang LJ, Liu SJ, Zhao QH,
Gao MM, Yi YH, Liao WP, Sun XF and Long YS: Alteration of Scn3a
expression is mediated via CpG methylation and MBD2 in mouse
hippocampus during postnatal development and seizure condition.
Biochim Biophys Acta. 1849:1–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morgan HD, Santos F, Green K, Dean W and
Reik W: Epigenetic reprogramming in mammals. Hum Mol Genet.
14:R47–R58. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gu Y, Zhang Z, Yin J, Ye J, Song Y, Liu H,
Xiong Y, Lu M, Zheng G and He Z: Epigenetic silencing of miR-493
increases the resistance to cisplatin in lung cancer by targeting
tongue cancer resistance-related protein 1 (TCRP1). J Exp Clin
Cancer Res. 36:1142017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dexheimer GM, Alves J, Reckziegel L,
Lazzaretti G and Abujamra AL: DNA methylation events as markers for
diagnosis and management of acute myeloid leukemia and
myelodysplastic syndrome. Dis Markers. 2017:54728932017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Seisenberger S, Peat JR, Hore TA, Santos
F, Dean W and Reik W: Reprogramming DNA methylation in the
mammalian life cycle: Building and breaking epigenetic barriers.
Philos Trans R Soc Lond B Biol Sci. 368:201103302013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Smallwood SA, Tomizawa S, Krueger F, Ruf
N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR and Kelsey
G: Dynamic CpG island methylation landscape in oocytes and
preimplantation embryos. Nat Genet. 43:811–814. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu B, Russanova VR, Gravina S, Hartley S,
Mullikin JC, Ignezweski A, Graham J, Segars JH, DeCherney AH and
Howard BH: DNA methylome and transcriptome sequencing in human
ovarian granulosa cells links age-related changes in gene
expression to gene body methylation and 3′-end GC density.
Oncotarget. 6:3627–3643. 2015.PubMed/NCBI
|
|
15
|
Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y,
Yan J, Ren X, Lin S, Li J, et al: The DNA methylation landscape of
human early embryos. Nature. 511:606–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang Z, Lin J, Dong H, Zheng X, Marjani
SL, Duan J, Ouyang Z, Chen J and Tian XC: DNA methylomes of bovine
gametes and in vivo produced preimplantation embryos. Biol
Reprod. 99:949–959. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y,
Yan J, Ren X, Lin S, Li J, et al: The DNA methylation landscape of
human early embryos. Nature. 511:606–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Down TA, Rakyan VK, Turner DJ, Flicek P,
Li H, Kulesha E, Gräf S, Johnson N, Herrero J, Tomazou EM, et al: A
Bayesian deconvolution strategy for immunoprecipitation-based DNA
methylome analysis. Nat Biotech. 26:779–785. 2008. View Article : Google Scholar
|
|
19
|
Cao XW, Lin K, Li CY and Yuan CW: A review
of WHO Laboratory Manual for the Examination and Processing of
Human Semen (5th edition). Zhonghua Nan Ke Xue. 17:1059–1063.
2011.(In Chinese). PubMed/NCBI
|
|
20
|
Yoshida K, Sekiguchi K, Mizuno N, Kasai K,
Sakai I, Sato H and Seta S: The modified method of two-step
differential extraction of sperm and vaginal epithelial cell DNA
from vaginal fluid mixed with semen. Forensic Sci Int. 72:25–33.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scacheri PC, Crawford GE and Davis S:
Statistics for ChIP-chip and DNase hypersensitivity experiments on
NimbleGen arrays. Methods Enzymol. 411:270–282. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Holliday R and Pugh JE: DNA modification
mechanisms and gene activity during development. Science.
187:226–232. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiao Y, Yu F, Pang L, Zhao H, Liu L, Zhang
G, Liu T, Zhang H, Fan H, Zhang Y, et al: MeSiC: a model-based
method for estimating 5 mC levels at single-CpG resolution from
MeDIP-seq. Sci Rep. 5:146992015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He S, Sun H, Lin L, Zhang Y, Chen J, Liang
L, Li Y, Zhang M, Yang X, Wang X, et al: Passive DNA demethylation
preferentially up-regulates pluripotency-related genes and
facilitates the generation of induced pluripotent stem cells. J
Biol Chem. 292:18542–18555. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vinci G, Buffat C, Simoncini S, Boubred F,
Ligi I, Dumont F, Le Bonniec B, Fournier T, Vaiman D, Dignat-George
F and Simeoni U: Gestational age-related patterns of AMOT
methylation are revealed in preterm infant endothelial progenitors.
PLoS One. 12:e01863212017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Beck S and Rakyan VK: The methylome:
Approaches for global DNA methylation profiling. Trends Genet.
24:231–237. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu P, Guo H, Ren Y, Hou Y, Dong J, Li R,
Lian Y, Fan X, Hu B, Gao Y, et al: Single-cell DNA methylome
sequencing of human preimplantation embryos. Nat Genet. 50:12–19.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miura F, Enomoto Y, Dairiki R and Ito T:
Amplification-free whole-genome bisulfite sequencing by
post-bisulfite adaptor tagging. Nucleic Acids Res. 40:e1362012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Taiwo O, Wilson GA, Morris T, Seisenberger
S, Reik W, Pearce D, Beck S and Butcher LM: Methylome analysis
using MeDIP-seq with low DNA concentrations. Nat Protoc. 7:617–636.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harrison A and Parle-McDermott A: DNA
methylation: A timeline of methods and applications. Front Genet.
2:742011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wardenaar R, Liu H, Colot V, Colomé-Tatché
M and Johannes F: Evaluation of MeDIP-chip in the context of
whole-genome bisulfite sequencing (WGBS-seq) in Arabidopsis.
Methods Mol Biol. 1067:203–224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cortijo S, Wardenaar R, Colomé-Tatché M,
Johannes F and Colot V: Genome-wide analysis of DNA methylation in
Arabidopsis using MeDIP-chip. Methods Mol Biol. 1112:125–149. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Seifert M, Cortijo S, Colomé-Tatché M,
Johannes F, Roudier F and Colot V: MeDIP-HMM: Genome-wide
identification of distinct DNA methylation states from high-density
tiling arrays. Bioinformatics. 28:2930–2939. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Smith ZD, Chan MM, Mikkelsen TS, Gu H,
Gnirke A, Regev A and Meissner A: A unique regulatory phase of DNA
methylation in the early mammalian embryo. Nature. 484:339–344.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Smith ZD, Chan MM, Humm KC, Karnik R,
Mekhoubad S, Regev A, Eggan K and Meissner A: DNA methylation
dynamics of the human preimplantation embryo. Nature. 511:611–615.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klose RJ and Bird AP: Genomic DNA
methylation: The mark and its mediators. Trends Biochem Sci.
31:89–97. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo F, Li X, Liang D, Li T, Zhu P, Guo H,
Wu X, Wen L, Gu TP, Hu B, et al: Active and passive demethylation
of male and female pronuclear DNA in the mammalian zygote. Cell
Stem Cell. 15:447–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hajkova P, Jeffries SJ, Lee C, Miller N,
Jackson SP and Surani MA: Genome-wide eprogramming in the mouse
germ line entails the base excision repair pathway. Science.
329:78–82. 2010. View Article : Google Scholar : PubMed/NCBI
|

















