Introduction
Ischemic stroke represents one of the major causes
of neuronal injury and disability, which may have increased
morbidity and high mortality rates during the next 20 years,
especially in developing countries (1). The overall cumulative risk of mortality
is 47% 5 years after an ischemic stroke (2). Reperfusion following ischemic stroke
may induce secondary brain damage, leading to neurological
dysfunction and cerebral endothelial injury, thereby increasing
cerebrovascular permeability and blood-brain barrier (BBB) leakage
(3). This is known as cerebral
ischemia/reperfusion (I/R) injury, which provides a notable
challenge for general surgeries and the treatment of ischemic
stroke (4). As such, there is an
urgent requirement to elucidate the development of the molecular
mechanisms underlying cerebral I/R injury and to identify novel
therapeutic strategies to improve clinical outcomes.
The Notch pathway is a highly conserved signaling
system that controls cellular self-renewal and survival during the
development of various tissues (5).
To date, 4 Notch receptors (Notch1-4) and 5 Notch ligands
(Jagged1/2 and Delta-like 1/3/4) have been identified to serve
important roles in the Notch signaling pathway (6). A series of proteolytic cleavages
initiated by the binding of Notch ligands to receptors are coupled
with the release and translocation of the intracellular Notch
receptor domain (NICD) to the nucleus, which activates the
transcription of Notch-targeted genes and regulates cell fate
decisions (7). In the central
nervous system (CNS), Notch is associated with the differentiation,
maturation, and functional maturation of neural progenitor cells
(8). The association of Notch with
I/R conditions in the cerebrum has previously been demonstrated,
and increased Notch1 expression has been observed during cerebral
ischemia (9–11). Furthermore, Notch has been reported
to be implicated in the regulation of endothelial cell
proliferation and vessel growth (12,13).
However, the underlying mechanism of Notch in cerebral I/R injury
associated with BBB dysfunction remains to be elucidated.
microRNAs (miRNAs or miRs) are a class of
endogenous, non-coding RNA molecules comprising 19–23 nucleotides,
which can modulate gene expression by binding with the
3′-untranslated region (UTR) of target mRNA molecules (14,15).
miRNAs serve important roles in physiological and pathologies
processes, which are associated with ischemic stroke (16–18). As
a key inflammation-related miRNA, miR-155 expression is
significantly altered by cerebral ischemia (19,20).
miR-155 has also been demonstrated to increase vascular endothelial
permeability (21,22). Therefore, understanding the role of
miR-155 and its mechanisms in cerebral I/R injury may provide an
informative approach to treating this disease.
The aim of the present study was to investigate the
role and molecular mechanism of miR-155 in the regulation of
cerebral I/R injury with middle cerebral artery occlusion (MCAO) in
mice, by assessing the function of BBB and the apoptosis index, and
measuring the expression of miR-155, Notch1, Jagged1 and hairy and
enhancer of split-1 (Hes1) in the ischemic cortex. These results
demonstrated a close association between Notch signaling and
miR-155 in cerebral I/R injury, indicating a potential target for
the treatment of ischemic stroke.
Materials and methods
Animals
A total of 120 male C57BL/6 mice were purchased from
the Experimental Animal Center of Guizhou Medical University
(Guiyang, China). Specific pathogen-free mice were housed under
steady state microenvironment conditions (temperature, 20±2°C; 12-h
light/dark cycle; 95% humidity), with ad libitum access to
food and water. All procedures were approved by the Animal Care and
Research Committee of The Affiliated Hospital of Guizhou Medical
University (Guiyang, China).
Experimental protocol
To evaluate the expression change of miR-155 during
I/R injury, 16 8-month-old C57BL/6 male mice (weight, 20–25 g) were
randomly divided into the following two groups (n=8 in each):
Sham-operated group (sham) and I/R group (Pre-IR). The Pre-IR group
was observed continuously for ≤24 h following I/R.
A conditional miR-155 knockout approach was
performed to reveal the role of the Notch signaling pathway in
ischemic brain injury. A miR-155 inhibitor (miR-155−/−)
and miR-155 mimics (miR-155+/+) were used. A total of
120 8-month-old C57BL/6 mice (weight, 20–25 g) were randomly
divided into the following six groups (n=20 in each): Sham, Pre-IR,
sham+miR-155 inhibitor (miR-155−/−sham), Pre-IR+miR-155
inhibitor (miR-155−/−Pre-IR), sham+miR-155 mimics
(miR-155+/+sham), Pre-IR+miR-155 mimics
(miR-155+/+Pre-IR). Mice in Sham groups were subjected
to surgical procedures without arterial occlusion, whereas mice in
the Pre-IR groups were subjected to MCAO.
Lentiviral transfection in mice
To modify the expression of miR-155 in the mouse
model, purified lentiviral particles containing
miR-155+/+ or miR-155−/− were obtained from
Shanghai GenePharma Co., Ltd. (Shanghai, China). The sequences were
as follows: miR-155+/+: 3′-UGGGCAUAGUCCUAAUCGUAAUU-5′;
miR-155−/−: 3′-UGCAUAUAAUGCUAAAGCAUUAA-5′; control
miRNA: 3′-UAAACAUGUACGCAUGCAUAGCU-5′. Prior to administration, mice
were anesthetized and fixed on a stereotactic frame, lentivirus
constructs (miR-155+/+, miR-155−/− and
scrambled control; 109 TU/ml) were mixed with the
cationic lipid Polybrene (4 µg/µl; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) and incubated at 37°C for 15 min. Subsequently,
each mouse was slowly administered 7 µl mixture over 20 min via
right intracerebroventricular injection. At 14 days following viral
vector injection, MCAO procedure was performed on these mice.
MCAO
The MCAO model was established in C57BL/6 mice
according to the methodology used in a previous study (23). Briefly, mice were anesthetized with
4% chloral hydrate (Sigma-Aldrich; Merck KGaA), and the left
common, internal (ICA) and external carotid arteries (ECA) were
carefully isolated. A 6-0 nylon suture was inserted into the ECA
stump, gently injected into the ICA and stopped at the opening of
the middle cerebral artery (MCA). The distance from the bifurcation
of ICA/ECA to MCA was ~10 mm. When the injection had been in place
for 90 min, nylon sutures were gently removed from the ICA and
reperfusion was performed (22).
Body temperature was maintained at 37°C during the surgical
procedure. Sham-operated mice received the same surgical procedure
without insertion of the nylon suture.
Evaluation of neurological scores
Following cerebral I/R injury, mice were assessed
for neurological deficits and scored by three blinded examiners as
described previously (24). Points
were awarded in the grading system as follows: 0, no deficit; 1,
forelimb weakness; 2, circling to affected side; 3, partial
paralysis on affected side; and 4, no spontaneous motor activity.
MCAO mice were allowed to recover for 24 h prior to evaluation.
Neurological scores were evaluated at 24, 48 and 72 h following
MCAO.
Staining with 2-3-5-triphenyl
terazolium chloride (TTC)
At 24 h following MCAO, five mice in each group were
anesthetized with intraperitoneal chloral hydrate (1 g/ml in
acetonitrile; 0.04 ml/100 g body weight; Sigma-Aldrich; Merck KGaA)
and sacrificed via decapitation. Mouse brains were harvested. The
sections were fixed in 10% formaldehyde for a minimum of 24 h at
4°C in the dark. Sections were then coronally sliced into 2-mm
slices. The slices were stained with 2% TCC (Sigma-Aldrich; Merck
KGaA) at 37°C for 20 min in the dark. The infarct brain area was
represented by white coloring, whereas normal brain tissues were
stained red. The infarct area was measured using MetaMorph software
(Molecular Devices, LLC, Sunnyvale, CA, USA). The infarct volume of
each slice was calculated as follows: V = t × (A1 + A2 + A3 + A4
+ A5) - (A1 + A2) × t/2 (A represents the infarct area of the
slice, t represents the slice thickness). The lesion volume is
presented as a volume percentage of the lesion compared with the
contralateral hemisphere as previously described (25).
Histological analysis
Brains from five mice in each group were fixed in 4%
paraformaldehyde overnight at 4°C, and subsequently washed with
0.9% saline and dehydrated. Following embedding the brain in
paraffin wax, 5-µm sections were cut and stained with hematoxylin
and eosin (HE) for 8 min at 37°C. Slides were viewed and imaged
under an Olympus light microscope (Olympus Corporation, Tokyo,
Japan) at a magnification of ×400.
Evans blue dye extravasation
method
Evans blue (2% in saline; 0.4 ml/100 g body weight;
Sigma-Aldrich; Merck KGaA) was injected into the jugular vein of
mice and allowed to circulate for 24 h. Subsequently, mice were
sacrificed and brains were quickly removed and weighed, then
homogenized in 2.5 ml PBS and centrifuged at 10,000 × g for 30 min
at 4°C. Evans Blue was measured using a spectrophotometer (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) at 610 nm.
Brain water content
Following sacrifice, five mouse brains in each group
were quickly removed and weighed (wet weight). The brains were then
dried at 120°C for 24 h and weighed again (dry weight). The
percentage of water content was calculated as follows: (wet
weight-dry weight)/wet weight ×100.
Nitric oxide (NO) release
NO production was measured using an NO assay kit
(cat. no. S0021; Beyotime Institute of Biotechnology, Haimen,
China) according to the manufacturer's protocol. The assay was
based on the Greiss test (26). Mice
were euthanized, brains (n=5) were quickly removed, and tissues
were homogenized in radioimmunoprecipitation assay lysis buffer
(Beyotime Institute of Biotechnology) and centrifuged at 15,000 × g
for 15 min at 4°C. Following centrifugation, 50 µl supernatant or
standard NaNO2 were mixed with 50 µl Griess reagent I
and 50 µl Griess reagent II in a 96-well plate for 10 min at 4°C.
Nitrite was measured using a spectrophotometer (Thermo Fisher
Scientific, Inc.) at 560 nm and quantified by the colorimetric MTT
assay (Sigma-Aldrich; Merck KGaA).
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) staining
Apoptotic cell death was detected using a TUNEL
staining kit (Roche Applied Science, Penzberg, Germany) according
to the manufacturer's instructions. Paraffin-embedded brain
sections were deparaffinized in dimethylbenzene, rehydrated with
gradient alcohol and digested with Proteinase K (20 µg/ml;
Sigma-Aldrich; Merck KGaA) for 30 min at 37°C. Following
pretreatment, sections were incubated in a solution containing
Biotinylated Nucleotide mix and Terminal Deoxynucleotidyl
Transferase at 37°C for 1 h. Antifade fluorescence mounting media
was added to the coverslip and the edges were sealed with nail
polish. Samples were observed and images were captured under
fluorescence microscopy. Five fields of view were observed at a
magnification of ×400.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from mouse brains (n=5)
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. Reverse
transcription was performed using universal primers and the
PrimeScript™ RT reagent kit (Takara Biotechnology Co., Ltd.,
Dalian, China) at 37°C for 15 min and 85°C for 5 sec. Transcripts
were quantified by qPCR using a BioRad CFX96 real-time PCR
detection system with SYBR-Green iQ™ Supermix (both from Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The PCR reaction was
performed at 95°C for 3 min, followed by 40 cycles of 95°C for 12
sec and 60°C for 40 sec. An internal control (GAPDH) was used to
normalize the expression of the target genes. The following primers
were used to amplify the gene fragments, and were available from
Genebank (https://www.ncbi.nlm.nih.gov/genbank/): miR-155,
forward, 5′-GCTTCGGTTAATGCTAATCGTG-3′ and reverse,
5′-CAGAGCAGGGTCCGAGGTA-3′; Notch1, forward,
5′-TCAATGTTCGAGGACCAGATG-3′ and reverse,
5′-TCACTGTTGCCTGTCTCAAG-3′; Hes1, forward,
5′-AGCCAACTGAAAACACCTGATT-3′ and reverse,
5′-GGACTTTATGATTAGCAGTGG-3′; and GAPDH, forward,
5′-CATGGCCTTCCGTGTTCCTA-3′ and reverse, 5′-CCTGCTTCACCACCTTCTTGA-3′
(Shanghai GenePharma Co., Ltd., Shanghai, China). The relative
quantification of the gene expression was calculated using the
comparative quantification cycle (Cq) method (27). Mean Cq values and deviations between
the duplicates were calculated; ΔCq=(Cq mRNA-Cq mRNA control), and
fold change=(2−ΔCq mRNA/2−ΔCq mRNA control).
RT-qPCR analysis of the mRNA levels of miR-155 was measured in
mouse brains at 6, 12 and 24 h following MCAO.
Western blotting
Mouse brains (n=5) were homogenized in RIPA lysis
buffer (Beyotime Institute of Biotechnology) by the BCA method, and
homogenates were centrifuged at 12,000 × g for 15 min at 4°C.
Supernatants were collected and stored at −80°C. Protein samples
(50 µg/lane) were separated by SDS-PAGE on 10% gels and proteins
were transferred onto polyvinylidene difluoride membranes.
Following the transfer, the membranes were blocked for 2 h with 5%
nonfat dry milk at room temperature. Membranes were incubated
overnight at 4°C, with the following primary antibodies:
Anti-endothelial NO synthase (eNOS; cat. no. 32027; 1:1,000),
anti-caspase3 (cat. no. 9662; 1:1,000), anti-Notch1 (cat. no. 3608;
1:1,000), anti-Hes1 (cat. no. 11988; 1:1,000), anti-β-Actin (cat.
no. 3700; 1:2,000; all Cell Signaling Technology, Inc., Danvers,
MA, USA) and anti-NICD (cat. no. ab8925; 1:1,000; Abcam, Cambridge,
UK). Following incubation with the primary antibodies, membranes
were washed with Tris-buffered saline containing 0.05% Tween-20
(TBST) 3 times, and subsequently incubated with appropriate
horseradish peroxidase-conjugated anti-rabbit secondary antibodies
lgG (cat. no. 14708; 1:2,000; Cell Signaling Technology, Inc.) at
room temperature for 2 h. Membranes were visualized using an
enhanced chemiluminescence assay kit (Merck KGaA). Protein bands
were digitally scanned and then analyzed using ImageJ software
(version 1.48; National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
Data are presented as the mean ± standard deviation
of at least three experiments. Statistical analyses were performed
using SPSS 22.0 (IBM Corp., Armonk, NY, USA). Differences among
groups were analyzed using by one-way analysis of variance with
post hoc Student-Newman-Keuls test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of cerebral I/R injury in
mice
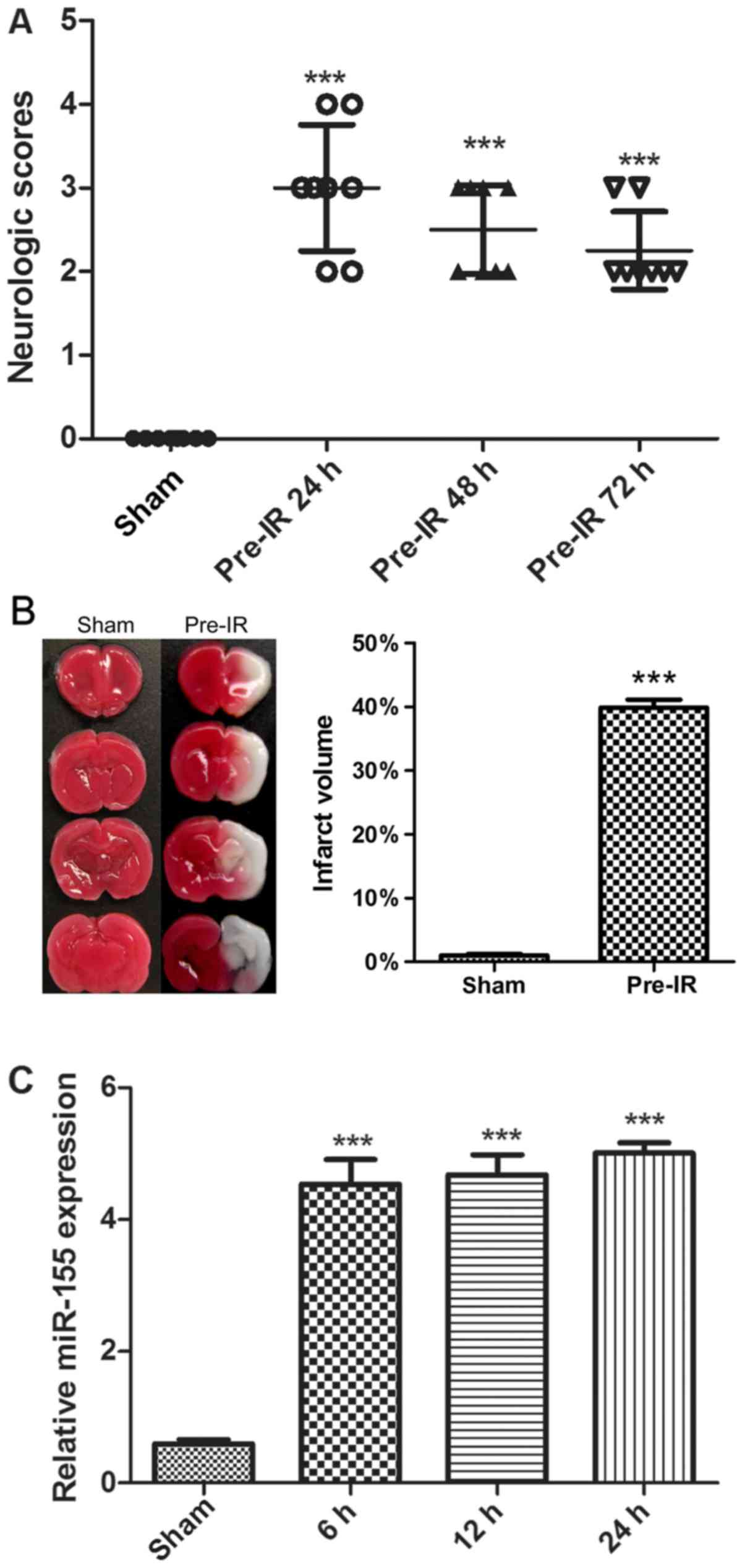
To determine the effects of cerebral I/R injury on
mice, neurological scores and TTC staining were performed. As
presented in Fig. 1A and B, there
was no notable neurological deficit and infarct exhibited by the
sham group. Compared with the sham group, the MCAO mice in the
Pre-IR group exhibited a significant increase in neurological
deficit and infarct (P<0.001). Furthermore, the neurological
deficit was alleviated over time.
To evaluate cerebral I/R injury on the regulation of
miR-155 expression, miR-155 mRNAs were analyzed via RT-qPCR. As
presented in Fig. 1C, cerebral I/R
injury significantly increased the synthesized levels of miR-155
mRNAs in brains, in comparison with the sham group (P<0.001).
The maximal expression of miR-155 was observed at 24 h.
Effects of miR-155 on BBB following
cerebral I/R injury in mice
To determine the effects of miR-155 on the brain
histopathology, HE staining was evaluated. As presented in Fig. 2A, the brain cortical tissue was
intact in the sham group. In accordance with this, the neurons were
integral and cellular structures were clear. However, the cortical
tissues exhibited congestion and edema, and the number of nerve
cells were markedly decreased in the Pre-IR group. In the
miR-155−/−Pre-IR group, neurons were arranged in neat
rows, and hyperemia and edema vanished. Intriguingly, the
histopathologic changes observed in the miR-155+/+Pre-IR
group appeared similar to those in the Pre-IR group, which
indicated that the cortical tissues in this group exhibited serious
hyperemia and edema accompanied by decreased cortical neurons.
 | Figure 2.Effects of miR-155 on brain
endothelial BBB following cerebral I/R injury in mice. (A)
Hematoxylin and eosin staining (magnification, ×40), (B) brain
water content and (C) Evans blue staining were quantified as
indications of BBB integrity at 24 h following MCAO (n=5). (D) NO
levels in brains were tested at 24 h following MCAO (n=5). (E)
Western blot analysis of the eNOS protein levels in brains at 24 h
following MCAO (n=5). Data are expressed as the mean ± standard
deviation. *P<0.05, **P<0.01, ***P<0.001. miR, microRNA;
BBB, blood-brain barrier; I/R, ischemia reperfusion; MCAO, middle
cerebral arterial occlusion; NO, nitric oxide; eNOS, endothelial NO
synthase; miR-155−/−, miR-155 inhibitor;
miR-155+/+, miR-155 mimics. |
To investigate the effects of miR-155 on BBB
permeability, Evans blue staining and brain water content were
evaluated. As presented in Fig. 2B and
C, the content of Evans blue and brain water exhibited a
significant increase in the Pre-IR group compared with the sham
group (P<0.001). However, miR-155−/− significantly
decreased the Evans blue and brain water levels in the
miR-155−/−Pre-IR group compared with the Pre-I/R group.
Compared with the miR-155−/−Pre-IR group, the content of
Evans blue and brain water in the miR-155+/+Pre-IR group
were significantly ameliorated (P<0.01).
To evaluate the effects of miR-155 on vascular
endothelial functions, NO content and the protein expression eNOS
were assayed. As presented in Fig. 2D
and E, the content of NO and the expression of eNOS in brains
were significantly decreased in the Pre-IR group, compared with the
sham group (P<0.001). However, miR-155−/−
significantly ameliorated NO and eNOS levels (P<0.001). Compared
with the miR-155−/−Pre-IR group, the content of NO and
the expression of eNOS in miR-155+/+Pre-IR group were
significantly decreased to levels similar to those of the Pre-IR
group (P<0.001).
Effects of miR-155 on apoptosis
following cerebral I/R injury in mice
As presented in Fig.
3A, apoptotic cells (green) were almost absent in ischemic area
of the sham group. The percentage of apoptotic cells was
significantly increased in ischemic areas following MCAO procedure
compared with the sham group (P<0.001). The percentage of TUNEL
positive cells was significantly decreased in the
miR-155−/−Pre-IR group compared with the Pre-I/R group
(P<0.001). Compared with the miR-155−/−Pre-IR group,
the TUNEL positive cells in the miR-155+/+Pre-IR group
were significantly ameliorated to levels similar to those of the
Pre-IR group (P<0.001).
As presented in Fig.
3B, the expression of caspase-3 in mouse brains was
significantly increased in the Pre-IR group compared with the sham
group (P<0.001). However, miR-155−/− significantly
decreased the caspase-3 levels in miR-155−/−Pre-IR group
compared with the Pre-I/R group (P<0.01). In comparison with the
miR-155−/−Pre-IR group, the caspase-3 level in the
miR-155+/+Pre-IR group was significantly ameliorated to
a level similar to that of the Pre-IR group (P<0.01).
Effects of miR-155 on the Notch
signaling pathway following cerebral I/R injury in mice
To investigate whether miR-155 serves a role in the
Notch signaling pathway, the expression of Notch1, NICD, Jagged1
and Hes1 were analyzed via western blotting. As presented in
Fig. 4A and B, the protein levels of
Notch1 were increased in increased in the Pre-IR group in
comparison with the sham group (P<0.001). However, compared with
the Pre-IR group, the levels of Notch1 in the
miR-155−/−Pre-IR group were significantly increased
(P<0.05). In comparison with the miR-155−/−Pre-IR
group, the expression of Notch1 in the miR-155+/+Pre-IR
group was significantly decreased to similar levels to the Pre-IR
group (P<0.05). As presented in Fig.
4A, C and D, changes in NICD and Hes1 levels were similar to
those of Notch1.
 | Figure 4.Effects of miR-155 on the Notch
signaling pathway following cerebral I/R injury in mice. (A)
Protein expression of the Notch pathway in brains at 24 h following
MCAO (n=5). Western blot analysis of Notch1 (B), NICD (C), and Hes1
(D) protein levels in brains. Reverse transcription-quantitative
polymerase chain reaction analysis of the (E) Notch1 and (F) Hes1
mRNA levels in brains at 24 h following MCAO (n=5). Data are
expressed as the mean ± standard deviation. *P<0.05,
**P<0.01, ***P<0.001. miR, microRNA; I/R, ischemia
reperfusion; MCAO, middle cerebral arterial occlusion; NICD,
intracellular Notch receptor domain; Hes1, hairy and enhancer of
split-1; miR-155−/−, miR-155 inhibitor;
miR-155+/+, miR-155 mimics. |
To investigate whether miR-155 has a role in the
Notch signaling pathway, the Notch1 and Hes1 mRNA levels were
analyzed via RT-qPCR. As presented in Fig. 4E, the mRNA levels of Notch1 was
significantly increased in the Pre-I/R group in comparison with the
sham group (P<0.001). However, compared with the Pre-IR group,
the mRNAs level of Notch1 in the miR-155−/−Pre-IR group
was significantly increased (P<0.001). In comparison with the
miR-155−/−Pre-IR group, the levels of Notch1 in
miR-155+/+Pre-IR group was signifcantly decreased
(P<0.001). As presented in Fig.
4F, the trend of Hes1 changes was similar to that of
Notch1.
Discussion
The CNS is a part of the nervous system that
contains a large number of microvascular cells, which permits a
close association between neurons and the endothelium (28). The BBB is a unique microvascular
structure in the brain, which helps maintain the homeostatic
microenvironment for normal neuronal functions and controls the
passage of material between blood and the brain. Cerebral I/R
injury states impair endothelial function, which may produce
vasogenic brain edema, hemorrhagic transformation and BBB leakage,
leading to neuronal apoptosis and neurological damage (29,30).
The present study demonstrated that the MCAO
procedure induced a cerebral I/R injury state with neuronal
apoptosis, and an increase in brain water and Evans blue content.
MCAO also induced an increase in infarct volume and TUNEL positive
cells with enhanced caspase-3 expression. The NO content in brains
and the expression of eNOS were found to be significantly decreased
in the Pre-IR group compared with the sham group. eNOS is
predominantly expressed in the vasculature, and catalyzes the amino
acid L-arginine conversion to NO, thus contributing to the
maintenance of endovascular homeostasis (31) and the regulation of cerebral blood
flow (32). eNOS polymorphisms are
accompanied by decreasing NO, which has been reported to be
associated with carotid atherosclerosis (33) or cerebral ischemia (34). This is consistent with the findings
of the present study. Previous studies suggested that deficiency in
endothelial NO and eNOS may cause cerebrovascular pathology and
neurological disease, which is a pathogenesis of cerebral ischemia
injury.
Notably, the present data also demonstrated that the
miR-155 mRNA levels were changed following MCAO procedure. miR-155,
the product of the B-cell integration cluster gene, was identified
as a critical miRNA during inflammation or cancer (35,36).
Previous studies have indicated that there is a relationship
between miR-155 and the endothelium. Liu et al (37) have reported miR-155 reduces umbilical
vein endothelial cells damage via regulating exogenous angiotensin
II. However, Huang et al (38) reported that the overexpression of
miR-155 contributed to inflammation-mediated glomerular endothelial
injury in diabetic nephropathy. In addition, Gama-Norton et
al (39) have demonstrated that
the Notch signal pathway controls cell fate in the hemogenic
endothelium. Ramasamy et al (12) have indicated that Notch signaling
promotes endothelial cell proliferation and vessel growth in
postnatal long bone. A previous study have also reported there is
an association between miR-155 and Notch signaling,
Notch/recombination signal binding protein immunoglobulin
κJ-signaling affects miR-155 in bone marrow endothelial cells,
leading to NF-κB activation and increased proinflammatory cytokine
production (40). It was therefore
hypothesized that the cerebral I/R induced neuronal apoptosis and
BBB damage may be associated with miR-155 and the Notch signaling
pathway. Therefore, a major aim of the present study was to
determine whether there is an association between miR-155 and the
Notch signaling pathway.
The results of the present study indicated that
deletion of miR-155 raised the expression of Notch1, NICD and Hes1,
thus activating the Notch signaling pathway. In addition, the
percentage of TUNEL-positive cells and caspase-3 levels were
decreased, with alleviated neuronal apoptosis. These findings
indicated that miR-155 may be an essential regulator of the Notch
signaling pathway.
Additionally, there were a number of limitations in
the present study. It is not fully clear yet if Notch1 is directly
targeted by miR-155, due to the absence of in vitro
experiments and detailed molecular mechanism research in the
present study. However, this limitation also provides a direction
for future research, and more work is needed to detect and explore
the mechanism underlying the miR155 effects on the Notch signaling
pathway.
Furthermore, it was hypothesized that miR-155
downregulates eNOS expression mainly through decreasing the Notch
pathway. The Notch pathway is important in controlling vascular
smooth muscle cells, and is also critical to vascular development,
repair, and remodeling (41–44). NICD may move into the
signal-receiving cell nucleus and regulates the expression of
downstream target genes, such as Hes1 (45). Yu et al (46) recently reported that the activation
of Notch1 and Hes1 is instrumental in withstanding myocardial I/R
injury. Tu et al (47)
previously demonstrated that the activated Notch signaling pathway
has been involved in the arteriovenous lesions in mice in
accordance with the present conjecture. The present findings
suggested that the activation of the Notch pathway upregulated the
eNOS expression and NO production and alleviated cerebral ischemic
injury, resulting in cerebrovascular pathology and neurological
disease.
The present study demonstrated that miR-155 was
involved in the negative regulation of NO production and eNOS
expression via the Notch signaling pathway. Deletion of miR-155
contributed to cerebral I/R injury-induced decreases of NO
production, eNOS expression and neuronal apoptosis through
upregulation of Notch1, NICD and Hes1. Overexpression of miR-155
aggravated BBB leakage and neuronal apoptosis, and downregulation
of Notch1, NICD and Hes1. These findings suggest that miR-155
serves an essential role in regulating the Notch signaling pathway
of the nervous system. Furthermore, these findings lay a foundation
for using miR-155 in the treatment of cerebral stroke.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
Guizhou Province Science and Technology Project (grant no. Qiankehe
Jichu [2016] 1126).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TJ constructed the middle cerebral artery occlusion
model and performed western blotting. SZ and XL performed the
histological analysis and statistical analysis. JS and TA conducted
the 2-3-5-triphenyl terazolium chloride staining and measured the
brain water content. XH and XP conducted the TUNEL staining and
reverse transcription-quantitative polymerase chain reaction
analysis. LW was responsible for the design of the study and
drafting the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures were approved by the Animal Care and
Research Committee of The Affiliated Hospital of Guizhou Medical
University (Guiyang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Donnan GA, Fisher M, Macleod M and Davis
SM: Stroke. Lancet. 371:1612–1623. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cabral NL, Nagel V, Conforto AB, Magalhaes
PS, Venancio VG, Safanelli J, Ibiapina F, Mazin S, França P,
Liberato RM, et al: High five-year mortality rates of ischemic
stroke subtypes: A prospective cohort study in Brazil. Int J
Stroke. Oct 9–2018.(Epub ahead of print). View Article : Google Scholar
|
|
3
|
Zhang HT, Zhang P, Gao Y, Li CL, Wang HJ,
Chen LC, Feng Y, Li RY, Li YL and Jiang CL: Early VEGF inhibition
attenuates blood-brain barrier disruption in ischemic rat brains by
regulating the expression of MMPs. Mol Med Rep. 15:57–64. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andersson ER, Sandberg R and Lendahl U:
Notch signaling: Simplicity in design, versatility in function.
Development. 138:3593–3612. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bray SJ: Notch signalling in context. Nat
Rev Mol Cell Biol. 17:722–735. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoon K and Gaiano N: Notch signaling in
the mammalian central nervous system: Insights from mouse mutants.
Nat Neurosci. 8:709–715. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun F, Mao X, Xie L, Ding M, Shao B and
Jin K: Notch1 signaling modulates neuronal progenitor activity in
the subventricular zone in response to aging and focal ischemia.
Aging Cell. 12:978–987. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
LeComte MD, Shimada IS, Sherwin C and
Spees JL: Notch1-STAT3-ETBR signaling axis controls reactive
astrocyte proliferation after brain injury. Proc Natl Acad Sci USA.
112:8726–8731. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pei H, Song X, Peng C, Tan Y, Li Y, Li X,
Ma S, Wang Q, Huang R, Yang D, et al: TNF-α inhibitor protects
against myocardial ischemia/reperfusion injury via Notch1-mediated
suppression of oxidative/nitrative stress. Free Radic Biol Med.
82:114–121. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramasamy SK, Kusumbe AP, Wang L and Adams
RH: Endothelial Notch activity promotes angiogenesis and
osteogenesis in bone. Nature. 507:376–380. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Patenaude A, Fuller M, Chang L, Wong F,
Paliouras G, Shaw R, Kyle AH, Umlandt P, Baker JH, Diaz E, et al:
Endothelial-specific Notch blockade inhibits vascular function and
tumor growth through an eNOS-dependent mechanism. Cancer Res.
74:2402–2411. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sepramaniam S, Tan JR, Tan KS, DeSilva DA,
Tavintharan S, Woon FP, Wang CW, Yong FL, Karolina DS, Kaur P, et
al: Circulating microRNAs as biomarkers of acute stroke. Int J Mol
Sci. 15:1418–1432. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu DZ, Tian Y, Ander BP, Xu H, Stamova
BS, Zhan X, Turner RJ, Jickling G and Sharp FR: Brain and blood
microRNA expression profiling of ischemic stroke, intracerebral
hemorrhage, and kainate seizures. J Cereb Blood Flow Metab.
30:92–101. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li SH, Su SY and Liu JL: Differential
regulation of microRNAs in patients with ischemic stroke. Curr
Neurovasc Res. 12:214–221. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Caballero-Garrido E, Pena-Philippides JC,
Lordkipanidze T, Bragin D, Yang Y, Erhardt EB and Roitbak T: In
vivo inhibition of miR-155 promotes recovery after experimental
mouse stroke. J Neurosci. 35:12446–12464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi GH, Ko KH, Kim JO, Kim J, Oh SH, Ha
IB, Cho KG, Kim OJ, Bae J and Kim NK: Association of miR-34a,
miR-130a, miR-150 and miR-155 polymorphisms with the risk of
ischemic stroke. Int J Mol Med. 38:345–356. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun HX, Zeng DY, Li RT, Pang RP, Yang H,
Hu YL, Zhang Q, Jiang Y, Huang LY, Tang YB, et al: Essential role
of microRNA-155 in regulating endothelium-dependent vasorelaxation
by targeting endothelial nitric oxide synthase. Hypertension.
60:1407–1414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cerutti C, Soblechero-Martin P, Wu D,
Lopez-Ramirez MA, de Vries H, Sharrack B, Male DK and Romero IA:
MicroRNA-155 contributes to shear-resistant leukocyte adhesion to
human brain endothelium in vitro. Fluids Barriers CNS. 13:82016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Huang J, Ma Y, Tang G, Liu Y, Chen
X, Zhang Z, Zeng L, Wang Y, Ouyang YB and Yang GY: MicroRNA-29b is
a therapeutic target in cerebral ischemia associated with aquaporin
4. J Cereb Blood Flow Metab. 35:1977–1984. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hara H, Friedlander RM, Gagliardini V,
Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J and Moskowitz
MA: Inhibition of interleukin 1beta converting enzyme family
proteases reduces ischemic and excitotoxic neuronal damage. Proc
Natl Acad Sci USA. 94:2007–2012. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li Y, Hua X, Hua F, Mao W, Wan L and Li S:
Are bone marrow regenerative cells ideal seed cells for the
treatment of cerebral ischemia? Neural Regen Res. 8:1201–1209.
2013.PubMed/NCBI
|
|
26
|
Xia P, Chen HY, Chen SF and Wang L,
Strappe PM, Yang HL, Zhou CH, Zhang X, Zhang YX, Ma LL and Wang L:
The stimulatory effects of eNOS/F92A-Cav1 on NO production and
angiogenesis in BMSCs. Biomed Pharmacother. 77:7–13. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
del Zoppo GJ and Mabuchi T: Cerebral
microvessel responses to focal ischemia. J Cereb Blood Flow Metab.
23:879–894. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Obermeier B, Daneman R and Ransohoff RM:
Development, maintenance and disruption of the blood-brain barrier.
Nat Med. 19:1584–1596. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moskowitz MA, Lo EH and Iadecola C: The
science of stroke: Mechanisms in search of treatments. Neuron.
67:181–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Srivastava K, Bath PM and Bayraktutan U:
Current therapeutic strategies to mitigate the eNOS dysfunction in
ischaemic stroke. Cell Mol Neurobiol. 32:319–336. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Milsom AB, Patel NS, Mazzon E, Tripatara
P, Storey A, Mota-Filipe H, Sepodes B, Webb AJ, Cuzzocrea S, Hobbs
AJ, et al: Role for endothelial nitric oxide synthase in
nitrite-induced protection against renal ischemia-reperfusion
injury in mice. Nitric Oxide. 22:141–148. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fatini C, Sofi F, Gensini F, Sticchi E,
Lari B, Pratesi G, Pulli R, Dorigo W, Pratesi C, Gensini GF and
Abbate R: Influence of eNOS gene polymorphisms on carotid
atherosclerosis. Eur J Vasc Endovasc Surg. 27:540–544. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Atochin DN, Clark J, Demchenko IT,
Moskowitz MA and Huang PL: Rapid cerebral ischemic preconditioning
in mice deficient in endothelial and neuronal nitric oxide
synthases. Stroke. 34:1299–1303. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Woodbury ME, Freilich RW, Cheng CJ, Asai
H, Ikezu S, Boucher JD, Slack F and Ikezu T: miR-155 Is essential
for inflammation-induced hippocampal neurogenic dysfunction. J
Neurosci. 35:9764–9781. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen S, Wang L, Fan J, Ye C, Dominguez D,
Zhang Y, Curiel TJ, Fang D, Kuzel TM and Zhang B: Host miR155
promotes tumor growth through a myeloid-derived suppressor
cell-dependent mechanism. Cancer Res. 75:519–531. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu T, Shen D, Xing S, Chen J, Yu Z, Wang
J, Wu B, Chi H, Zhao H, Liang Z and Chen C: Attenuation of
exogenous angiotensin II stress-induced damage and apoptosis in
human vascular endothelial cells via microRNA-155 expression. Int J
Mol Med. 31:188–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang Y, Liu Y, Li L, Su B, Yang L, Fan W,
Yin Q, Chen L, Cui T, Zhang J, et al: Involvement of
inflammation-related miR-155 and miR-146a in diabetic nephropathy:
Implications for glomerular endothelial injury. BMC Nephrol.
15:1422014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gama-Norton L, Ferrando E, Ruiz-Herguido
C, Liu Z, Guiu J, Islam AB, Lee SU, Yan M, Guidos CJ, López-Bigas
N, et al: Notch signal strength controls cell fate in the
haemogenic endothelium. Nat Commun. 6:85102015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang L, Zhang H, Rodriguez S, Cao L,
Parish J, Mumaw C, Zollman A, Kamoka MM, Mu J, Chen DZ, et al:
Notch-dependent repression of miR-155 in the bone marrow niche
regulates hematopoiesis in an NF-κB-dependent manner. Cell Stem
Cell. 15:51–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kurpinski K, Lam H, Chu J, Wang A, Kim A,
Tsay E, Agrawal S, Schaffer DV and Li S: Transforming growth
factor-beta and notch signaling mediate stem cell differentiation
into smooth muscle cells. Stem Cells. 28:734–742. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hofmann JJ and Iruela-Arispe ML: Notch
signaling in blood vessels: Who is talking to whom about what? Circ
Res. 100:1556–1568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Holderfield MT and Hughes CC: Crosstalk
between vascular endothelial growth factor, notch, and transforming
growth factor-beta in vascular morphogenesis. Circ Res.
102:637–652. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zou S, Ren P, Nguyen M, Coselli JS, Shen
YH and LeMaire SA: Notch signaling in descending thoracic aortic
aneurysm and dissection. PLoS One. 7:e528332012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yan B, Raben N and Plotz P: The human acid
alpha-glucosidase gene is a novel target of the Notch-1/Hes-1
signaling pathway. J Biol Chem. 277:29760–29764. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu L, Liang H, Lu Z, Zhao G, Zhai M, Yang
Y, Yang J, Yi D, Chen W, Wang X, et al: Membrane receptor-dependent
Notch1/Hes1 activation by melatonin protects against myocardial
ischemia-reperfusion injury: In vivo and in vitro studies. J Pineal
Res. 59:420–433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tu J, Li Y, Hu Z and Chen Z: Radiosurgery
inhibition of the Notch signaling pathway in a rat model of
arteriovenous malformations. J Neurosurg. 120:1385–1396. 2014.
View Article : Google Scholar : PubMed/NCBI
|