Introduction
Mortality and morbidity rates as a result of cardiac
arrest (CA) remain high despite steady advances in therapeutic
approaches over the past decades (1). Post-resuscitation neurological
dysfunction is a major cause of mortality in patients following
successful cardiopulmonary resuscitation (CPR) (2,3).
Therefore, it is vital to research strategies to protect
neurological function in patients following CA.
Although CA can lead to ischemia/reperfusion (I/R)
injury in every organ in the body, it is especially detrimental to
the brain and the heart. The principal pathological cause of organ
damage is excessive apoptosis and autophagy (4,5), the
extent of which has been found to correlate with poor neurological
prognosis (6–8). A number of studies have demonstrated
that hypothermia can attenuate neurological damage following I/R
injury by reducing neural apoptosis and preventing autophagy
overactivation (4,9,10).
Therefore, it has been recommended as a potential treatment in
patients who were successfully resuscitated following CA (11).
Apoptosis is a form of cell death that is mediated
by several proteins, including Bax, Bcl-2, 70 kDa heat shock
protein (HSP70) and cysteinyl aspartate specific proteinase
(Caspase-3) (12,13). Bax and Bcl-2 are members of the Bcl-2
family of molecular protein factors that can reflect the extent of
cellular apoptosis (14); whereas
HSP70 is a highly conserved molecular chaperone that serves an
important role in cellular adaptation to stress, a requisite for
cell survival (15). Lastly,
Caspase-3 is a commonly probed apoptosis marker, as it takes part
in the execution-phase of the apoptotic pathway (13,16,17).
Autophagy is the primary mechanism by which
cytosolic proteins and organelles are degraded (18). However, excessive autophagy
activation in neurocytes damaged by I/R can lead to non-apoptotic
cell death (19,20), which is called autoghagic programmed
cell death. Beclin1 activates critical steps in autophagy,
including the formation and maturation of autophagosomes (21,22). In
addition, Bax and Bcl-2 have also been demonstrated to regulate
autophagy (12). In fact, it has
been well documented that the crosstalk between autophagy and
apoptosis pathways is mediated at least in part by the functional
and structural interaction between Beclin1 and Bcl-2 (23).
Dexmedetomidine (Dex) is a highly selective
α2-adrenergic receptor agonist that is commonly used as
a sedative and analgesic in the intensive care unit (24); it has been reported to display
synergistic effects with hypothermia (25,26).
Consequently, based on previous observations that hypothermia can
alleviate neuronal damage following CA, it was hypothesized in the
present study that Dex administration after CA may result in
improved neural function and reduced apoptosis and autophagy. The
present study also aimed to investigate the possible mechanism by
which Dex improves cognitive function.
Materials and methods
Animal preparation and Experimental
protocol
All operations and euthanasia were performed under
anesthesia induced by an intraperitoneal (i.p.) injection of 3.6%
chloral hydrate solution (3.6 g chloral hydrate crystal diluted
with 100 ml distilled water) at a concentration of 360 mg/kg, and
the best efforts were made to minimize animal suffering.
A total of 72, male Sprague-Dawley rats [age, 17 and
18 weeks; weight, 430±45 g; JOINN Laboratories (China) Co., Ltd.]
were recruited successfully. They were provided food and water
ad libitum, and housed at 25°C, with freshly ventilator
delivered atmosphere and a 12 h light/dark cycle. The procedure of
animal CA and CPR were performed as described previously (4). Endotracheal intubation was performed
prior to left/right femoral arteriovenous catheterization. Animals
were ventilated with a volume-controlled ventilator (Institute of
Cardiopulmonary Cerebral Resuscitation, Guangdong, China), with a
tidal volume of 6 ml/kg, a fraction of inspired oxygen
(FiO2) of 0.21, and a ventilation rate of 100 breaths
per min. Continuous arterial blood pressure was monitored via an
intra-arterial pressure sensor suite (Philips Medical Systems,
Inc.) and an electrocardiogram (Anhui Zhenghua Biological
Instrument Equipment Co., Ltd.). Baseline arterial blood gas
analysis was performed 10 min after the surgical procedure. An
equal volume (0.5 ml) of normal saline was supplemented into the
venous catheter immediately after arterial blood collection. CA was
induced in the animals by reducing mechanical ventilation and
occluding the tracheal catheter. For CPR, external chest
compression (250 bpm) and mechanical ventilation (6 ml/kg and 100
bpm) was performed after a period of 5 min under systolic blood
pressure <25 mmHg. After 10 sec of CPR all rats were
administered a bolus injection of epinephrine (10 µg/kg, diluted to
0.5 ml) in the right femoral vein. In addition to epinephrine,
before CPR execution, one group of rats received an i.p. injection
of Dex (50 µg/kg (27), diluted to
0.5 ml; D group) whereas another group received an i.p. injection
of 0.9% normal saline (0.5 ml; N group). ROSC achievement was
defined as the presence of an autonomic cardiac rhythm and a mean
arterial blood pressure >60 mmHg. At this point, chest
compressions were stopped, a FiO2 of 100% was maintained
for 15 min after ROSC, decreased to 45% for a further 15 min and
then decreased to 21% until the animal exhibited spontaneous
respirations. Resuscitation procedures were terminated if animals
were unresponsive to CPR for 10 min. After 2 h of monitoring animal
vital signs, rats returned to consciousness, were placed into a
thermostabilized (25°C) cage for rehabilitation and euthanized
either 12 (N-12, n=15; D-12, n=15) or 24 h (N-24, n=14; D-24, n=13)
later. A total of 9 rats died during the recovery period in the
cage due to airway edema and sputum obstruction, which was
certified following autopsy. Rats belonging to the blank control
group (BC; n=6) received identical surgical and anesthetic
treatments as the experimental rats with the exception of CA and
CPR.
Critical parameters, including core (rectal)
temperature and blood pressure were monitored continuously for 120
min (timed from the first sign of ROSC) after CPR until the rats
regained consciousness. Within the same timeframe, arterial blood
gas was also analyzed 30 and 120 min after CPR.
Neurological deficit scores (NDS)
NDS is a common procedure for evaluating
neurological dysfunction by using a hierarchical scoring system
based on respiratory pattern, consciousness, sensory and motor
function and behavioral responses (4). Every animal was evaluated 12 or 24 h
following return of spontaneous circulation (ROSC). The NDS items
were collected by investigators that have been specially trained,
who were blinded to the experimental groups. Individual scores were
subsequently combined to produce the final NDS score. A score of 80
is considered normal whereas a score of zero is considered brain
dead (28).
Collection of rat hippocampal
tissues
After the NDS scores were obtained, rats were
anesthetized using a combination of an i.p. injection of 3.6%
chloral hydrate solution at 360 mg/kg and an intracardial injection
of 500 mg/kg 10% potassium chloride solution. The hippocampus was
rapidly removed from the cerebrum of the rat. The left half was
snap frozen using liquid nitrogen before storage at −80°C to be
used later for western blot analysis of Bcl-2, Bax, HSP70 and
Beclin1 protein expression. The right half of the hippocampus was
fixed in 4% paraformaldehyde for 24 h at 4°C and subsequently
embedded in paraffin for analysis using histology techniques.
Western blot analysis of Bcl-2, Bax,
HSP70 and Beclin1 expression in the rat hippocampus
Hippocampal tissue protein was extracted with 400 µl
RIPA and 4 µl PMSF (Beyotime Institute of Biotechnology). Samples
were then homogenized on the ice and centrifuged in a refrigerated
centrifuge (12,000 × g for 10 min at 4°C). Protein concentration in
the supernatant from hippocampal homogenate was determined using a
Bicinchoninic Acid Assay kit (Beyotime Institute of Biotechnology).
A total of 50 µg of protein obtained from the hippocampus were
separated by 10 or 12% SDS-PAGE before transferal to a PVDF
membrane. Each membrane was blocked in 5% non-fat milk diluted in
TBS supplemented with 0.1% Tween-20 (TBST) for 1 h at 4°C. The
membranes were then incubated overnight at 4°C with primary
antibodies against Bcl-2 (1:1,000; cat. no. ab196495), Bax
(1:1,000; cat. no. ab32503), HSP70 (1:1,000; cat. no. ab137680),
Beclin1 (1:1,000; cat. no. ab62557) or Glyceraldehyde-3-phosphate
dehydrogenase (GAPDH; 1:800; cat. no. ab9485); all primary
antibodies applied for western blot analysis were purchased from
Abcam. The following day, the membranes were rinsed with TBS-T and
subsequently incubated with a biotin-conjugated goat anti-rabbit
IgG secondary antibody (1:5,000; cat. no. ab222773; Abcam) at room
temperature for 1 h. Protein bands were visualized using an ECL
reagent (Biological Industries Israel Beit Haemek Ltd.).
Densitometric analysis was performed using Quantity One 1-D
analysis software (version 4.6.9; Bio-Rad Laboratories, Inc.); the
protein levels of Bcl-2, Bax, HSP70 and Beclin1 were all normalized
to GAPDH.
Immunohistochemistry analysis of
Caspase-3
Paraffin-embedded hippocampal tissue was cut into 5
µm sections using a sledge microtome. The tissue was deparaffinized
using xylene and rehydrated in a series of descending
concentrations of ethanol. Antigen retrieval was completed by
boiling in citric acid buffer (10 mM, pH 6.0) at 95°C for 15 min.
0.5% Triton X-100 (Beyotime Institute of Biotechnology) was used
for membrane permeabilization. The slides were blocked at 37°C for
2 h in 5% bovine serum albumin (BSA; Beyotime Institute of
Biotechnology) diluted in TBS and subsequently incubated in
anti-caspase-3 antibody (1:100; cat. no. ab4051; Abcam) diluted in
TBS supplemented with 5% BSA overnight at 4°C. The following day,
the sections were rinsed in TBS prior to incubation with a
biotin-conjugated goat anti-rabbit secondary antibody (1:500; cat.
no. ab222773) for 1 h at 37°C. The secondary antibody was washed
off using TBS and the slides were incubated under 37°C for 5–10 min
in a solution containing 0.02% diaminobenzidine (DAB) and 0.01%
H2O2 and then counterstained using
hematoxylin and eosin (H&E) at 37°C for 10 min. The slides were
imaged using a light microscope (Olympus BX53; Olympus
Corporation). Cells exhibiting brown staining were considered
Caspase-3 positive and were counted using the Leica software
analysis system (Q550CW; Leica Microsystems, Inc). Each group was
observed in four different fields of view; 100 cells were observed
in the field of vision and then caspase-3 positive cells were
calculated. Counting was performed by a specialized pathologist who
was blind to the experimental groups.
Transmission electron microscopy
(TEM)
The paraffin-embedded tissues were also used for
histological analysis using TEM. This tissue was sliced using a
microtome into ultra-thin sections (~40–50 nm). The sections were
then stained with 2.0% (w/v) lead citrate at 4°C for 10 min and
evaluated using a Hitachi H-600 transmission electron microscope
(Hitachi, Ltd.) in a blinded fashion. For every section, four
different TEM micrographs each representing independent fields of
view were analyzed.
Statistical analysis
All experimental data are presented as the mean ±
SD. A t-test was used to compare the means between the two groups.
Multiple-comparison analyses performed in this study were confirmed
using Levene's test for the equality of variances. A one-way ANOVA
was applied to assess holistic differences between groups for each
variable, and Bonferroni post-hoc test was used for multiple
comparisons. The software package used to perform the analysis was
IBM SPSS Statistics 22.0 (IBM Corp.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Animal hemodynamic, physiological, and
resuscitation data
The experimental scheme of animal treatment for the
present study is detailed in Fig. 1.
No statistically significant differences were observed in
operation, hemodynamic stabilization, asphyxia, CA and CPR time
between the resuscitation groups (Table
I). In addition, no significant differences were identified
between the groups with regards to the respiration rate, mean
aortic blood pressure, arterial blood pH, oxygen and carbon dioxide
partial pressure, oxygen saturation and lactic acid during
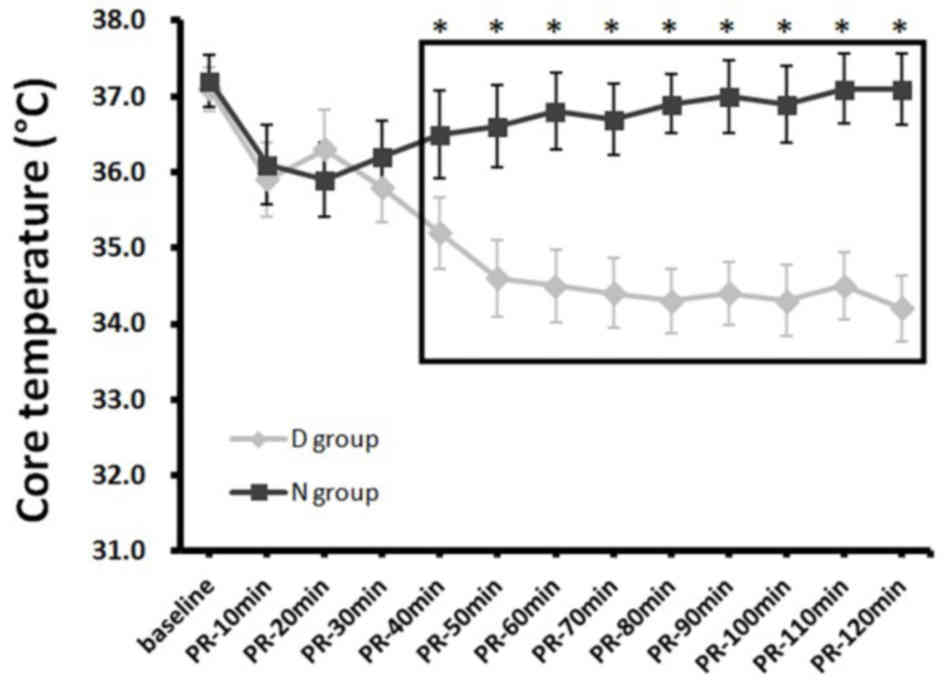
baseline, at either 30 min or 2 h after ROSC (Table II). Core body temperatures and heart
rates in the D group decreased over time, and began to exhibit
statistically significant reductions compared with the N group from
40 and 80 min post-ROSC, respectively, up to post-ROSC 120 min when
the analyses ended (both P<0.05; Figs. 2 and 3; Table
II).
 | Table I.Cardiac arrest time and associated
resuscitation data. |
Table I.
Cardiac arrest time and associated
resuscitation data.
| Group | Valid rats (n) | Time of operation
(sec) | Time of
stabilization for baseline (sec) | Time from asphyxia
to SBP <25 mmHg (sec) | Time of SBP <25
mmHg (sec) | Time from
resuscitation to ROSC (sec) |
|---|
| N | 29 | 485.61±62.47 | 589.24±15.72 | 221.26±36.43 | 272.34±36.75 | 86.41±32.67 |
| D | 28 | 505.73±58.32 | 596.12±12.67 | 234.82±32.48 | 268.28±38.74 | 89.64±29.53 |
| Table II.Animal physiological data and
arterial blood gas analysis. |
Table II.
Animal physiological data and
arterial blood gas analysis.
| A, N (n=29) |
|---|
|
|---|
| Timepoint | Tc
(°C) | HR (bpm) | RR (bpm) | MAP (mmHg) | pH | PaO2
(mmHg) | PaCO2
(mmHg) | SO2c
(%) | Lac (mg/dl) |
|---|
| Baseline | 37.25±0.48 | 262.84±39.42 | 79.87±2.81 | 92.35±25.52 | 7.37±0.07 | 95.43±18.71 | 40.35±5.41 | 95.24±6.45 | 1.2±1.17 |
| PR-0.5 h | 36.09±0.47 | 241.24±47.41 | 78.96±0.68 | 65.38±12.84 | 7.21±0.13 | 73.54±21.75 | 55.83±17.68 | 90.40±9.82 | 7.1±2.43 |
| PR-2 h | 36.85±0.53 | 251.47±51.84 | 92.42±13.24 | 89.49±26.75 | 7.28±0.09 | 86.68±24.24 | 47.37±8.54 | 92.86±8.23 | 3.6±1.76 |
|
| B, D
(n=28) |
|
|
Timepoint | Tc
(°C) | HR
(bpm) | RR
(bpm) | MAP
(mmHg) | pH | PaO2
(mmHg) | PaCO2
(mmHg) | SO2c
(%) | Lac
(mg/dl) |
|
| Baseline | 37.13±0.39 | 258.46±42.43 | 77.65±2.51 | 99.50±21.38 | 7.38±0.03 | 97.64±15.61 | 41.86±6.23 | 96.13±3.46 | 1.5±0.99 |
| PR-0.5 h | 35.75±0.41 | 229.28±42.37 | 79.86±0.67 | 61.12±13.58 | 7.25±0.12 | 71.46±19.73 | 52.16±13.26 | 91.53±7.43 | 7.2±1.92 |
| PR-2 h |
34.12±0.35a |
212.48±36.53a | 88.76±11.47 | 85.83±21.44 | 7.30±0.08 | 88.47±18.31 | 44.49±7.64 | 93.43±9.37 | 3.2±1.18 |
Neurological outcome
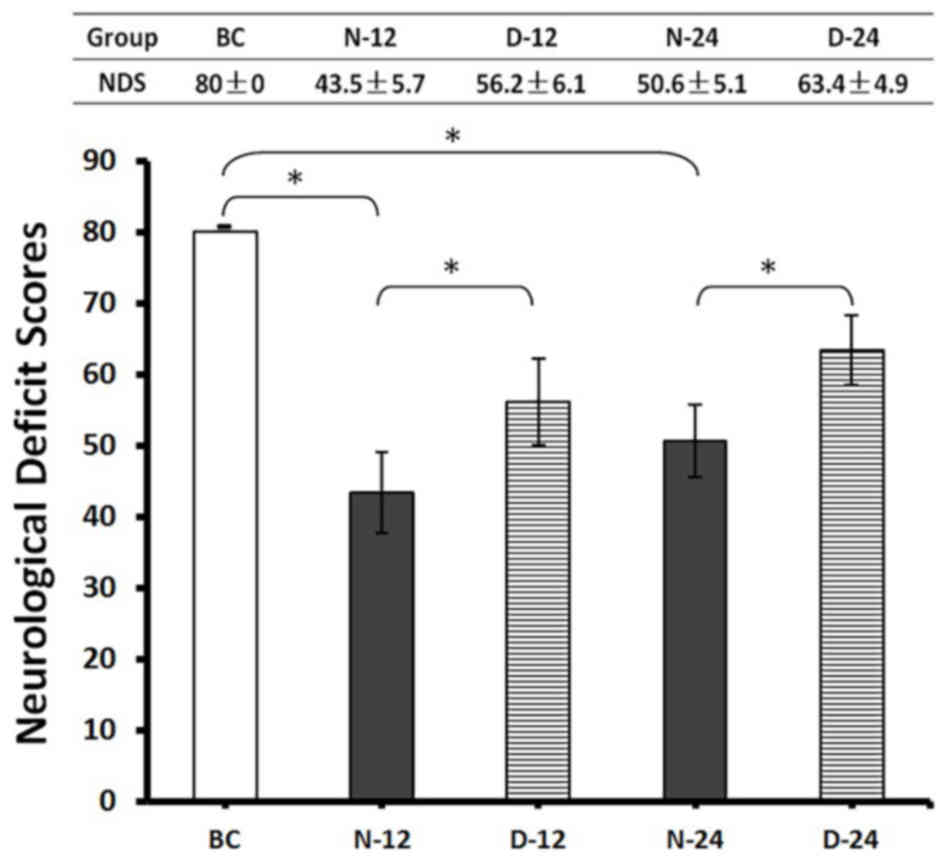
NDS was determined before the rats were euthanized
at 12 or 24 h post-ROSC. The mean NDS value of rats in the N-12
(43.5±5.7) and N-24 (50.6±5.1) groups was significantly lower
compared with the BC group (80.0±0.0; P<0.05; Fig. 4). However, post-ROSC NDS was higher
in the D-12 (56.2±6.1) and D-24 (63.4±4.9) groups compared with
their respective N-12 and N-24 timepoints (P<0.05; Fig. 4). These findings suggested that the
use of Dex post-ROSC in rats improved neurological outcomes after
CA compared with untreated animals.
Intracellular Bax, Bcl-2, HSP70 and
Beclin1 protein expression levels in the hippocampus
Bax expression was found to be greater in the N-12
(0.024±0.002) and N-24 (0.022±0.006) groups post-ROSC compared with
the BC group (0.003±0.000) (P<0.05; Fig. 5A). In addition, Bax expression was
significantly lower in the D-12 (0.017±0.001) and D-24
(0.006±0.004) groups post-ROSC compared with the respective N-12
and N-24 timepoints (P<0.05; Fig.
5A). Bcl-2 expression was significantly reduced in the N-12
(0.019±0.001) and N-24 (0.014±0.001) groups post-ROSC compared with
the BC group (0.022±0.000) (P<0.05; Fig. 5B). However, Bcl-2 expression was
significantly higher in the D-12 (0.025±0.001) and D-24
(0.024±0.006) groups post-ROSC compared with their respective N-12
and N-24 timepoints (P<0.05; Fig.
5B). HSP70 expression was higher in the N-12 (0.45±0.07) and
N-24 (0.41±0.05) groups post-ROSC compared with the BC group
(0.32±0.00) (P<0.05; Fig. 5C). In
addition, HSP70 expression was higher in the D-12 (0.56±0.10) and
D-24 (0.54±0.06) groups compared with the respective N-12 and N-24
timepoints (P<0.05; Fig. 5C).
Beclin1 expression was higher in the N-12 (0.43±0.05) and N-24
(0.39±0.04) groups post-ROSC compared with the BC group (0.15±0.00)
(P<0.05; Fig. 5D). However,
Beclin1 expression was lower in the D-12 (0.31±0.06) and D-24
(0.25±0.03) groups post-ROSC compared with their respective N-12
and N-24 timepoints (P<0.05; Fig.
5D).
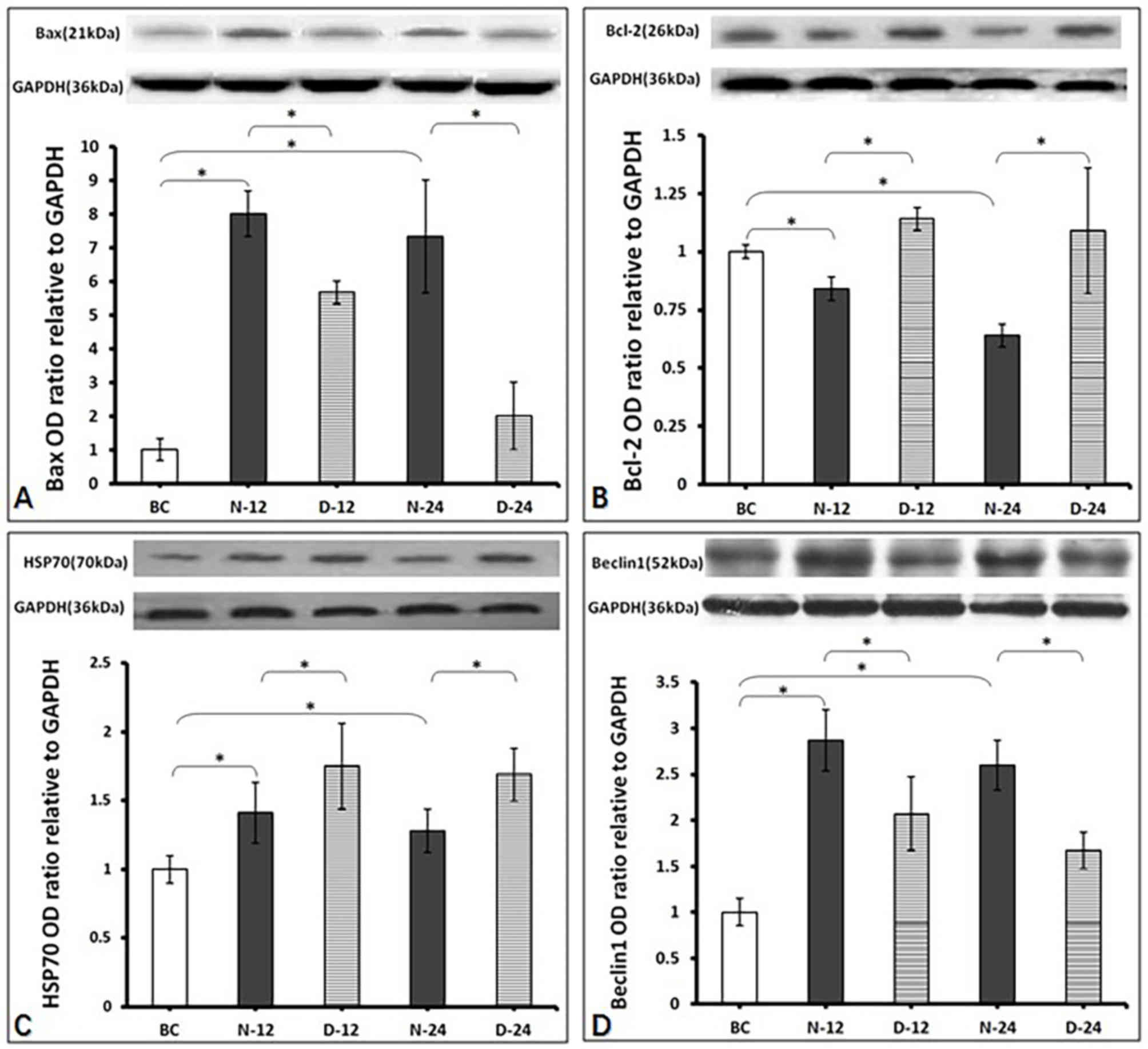 | Figure 5.Western blotting results. All data
values were normalized to the values of BC group (as level ‘1’).
(A) Measurement of Bax protein. The expression levels were in the
rat hippocampus from each treatment group 12 and 24 h after ROSC,
in the presence of normal saline or dexmedetomidine. *P<0.05.
BC, blank control group; D-12, dexmedetomidine group 12 h
post-ROSC; D-24, dexmedetomidine group 24 h post-ROSC; N-12, normal
saline group 12 h post-ROSC; N-24, normal saline group 24 h
post-ROSC; ROSC, return of spontaneous circulation. (B) Measurement
of Bcl-2 protein. The expression levels were in the rat hippocampus
from each treatment group 12 and 24 h after ROSC, in the presence
of normal saline or dexmedetomidine. *P<0.05. BC, blank control
group; D-12, dexmedetomidine group 12 h post-ROSC; D-24,
dexmedetomidine group 24 h post-ROSC; N-12, normal saline group 12
h post-ROSC; N-24, normal saline group 24 h post-ROSC; ROSC, return
of spontaneous circulation. (C) Measurement of HSP70 protein. The
expression levels were in the rat hippocampus from each treatment
group 12 and 24 h after ROSC, in the presence of normal saline or
dexmedetomidine. *P<0.05. BC, blank control group; D-12,
dexmedetomidine group 12 h post-ROSC; D-24, dexmedetomidine group
24 h post-ROSC; HSP70, heat shock protein 70 kDa; N-12, normal
saline group post-ROSC 12 h; N-24, normal saline group 24 h
post-ROSC; ROSC, return of spontaneous circulation. (D) Measurement
of Beclin1 protein. The expression levels were in the rat
hippocampus from each treatment group 12 and 24 h after ROSC, in
the presence of normal saline or dexmedetomidine. *P<0.05. BC,
blank control group; D-12, dexmedetomidine group 12 h post-ROSC;
D-24, dexmedetomidine group 24 h post-ROSC; N-12, normal saline
group 12 h post-ROSC; N-24, normal saline group 24 h post-ROSC;
ROSC, return of spontaneous circulation. |
Measurement of apoptosis in the rat
hippocampus
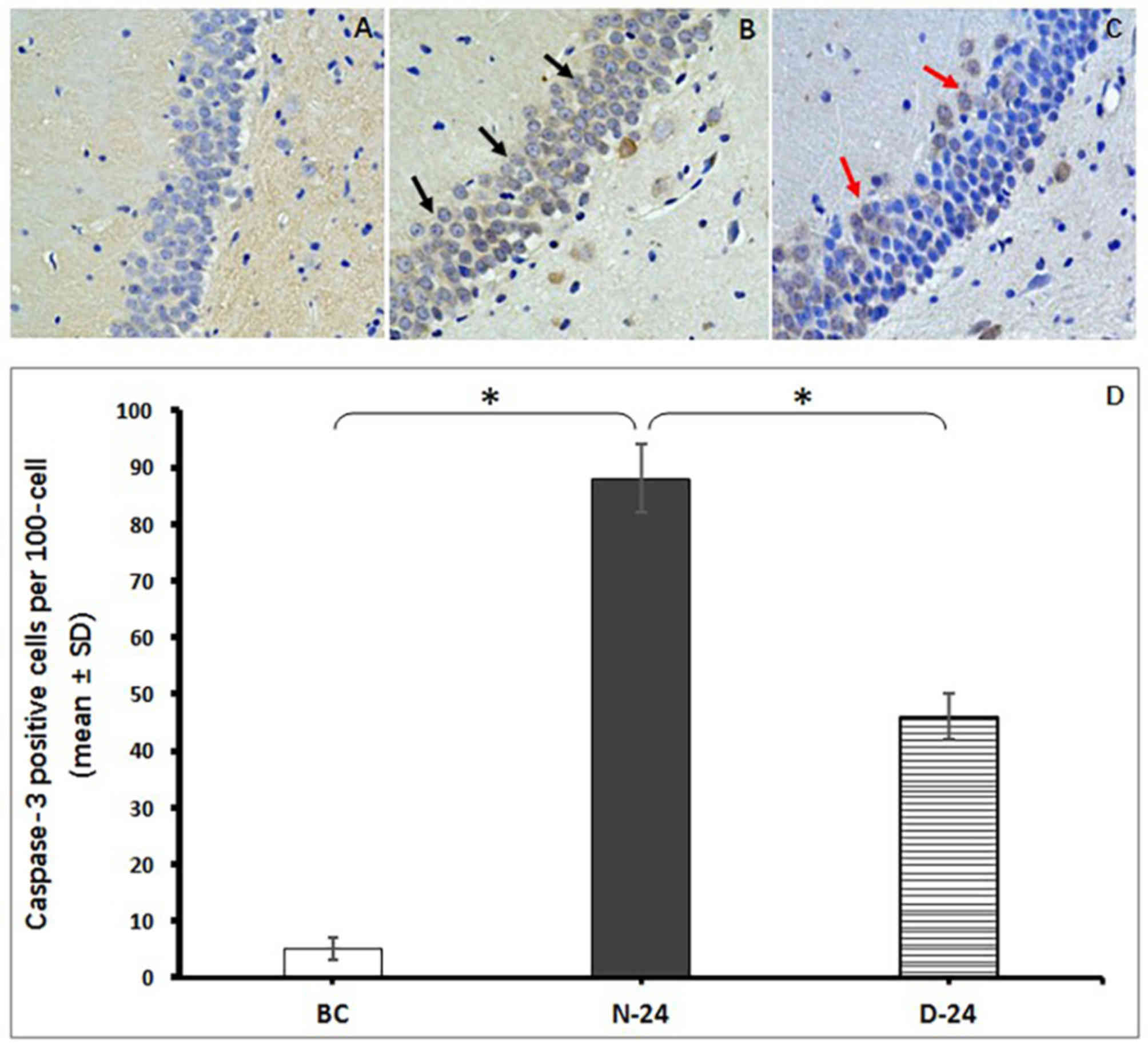
Neuronal apoptosis was determined by counting the
number of Caspase-3-positive staining cells located in the CA1
region of the hippocampus. The intense staining of surrounding
tissues was impacted by the time of DBA coloration and the brown
staining inside cells was defined as apoptosis-positive. The number
of apoptotic neurons were demonstrated to be more abundant in the
N-24 group compared with the BC group (P<0.05; Fig. 6). However, fewer hippocampal neurons
were stained positive for Caspase-3 in the D-24 group compared with
N-24 group (P<0.05; Fig. 6).
Histological and ultrastructural
analysis in the rat hippocampus
Histological analysis of hippocampal tissues from
the three groups was carried out using H&E staining 24 h
post-ROSC; following which the nuclei of cells located in the CA1
region of the hippocampus were examined. Compared with the distinct
shape and content of neurocytes from BC group (Fig. 7A; white arrows), cells from the N-24
group exhibited faint nuclear shapes with compact cytoplasm
(Fig. 7B; yellow arrows), which
indicated that hippocampal neurons were damaged after I/R. Cells
from the D-24 group hippocampus displayed visible nuclei and
distinct cytoplasm (Fig. 7C; green
arrows) compared with N group, which suggested that Dex treatment
reduced neural damage following I/R.
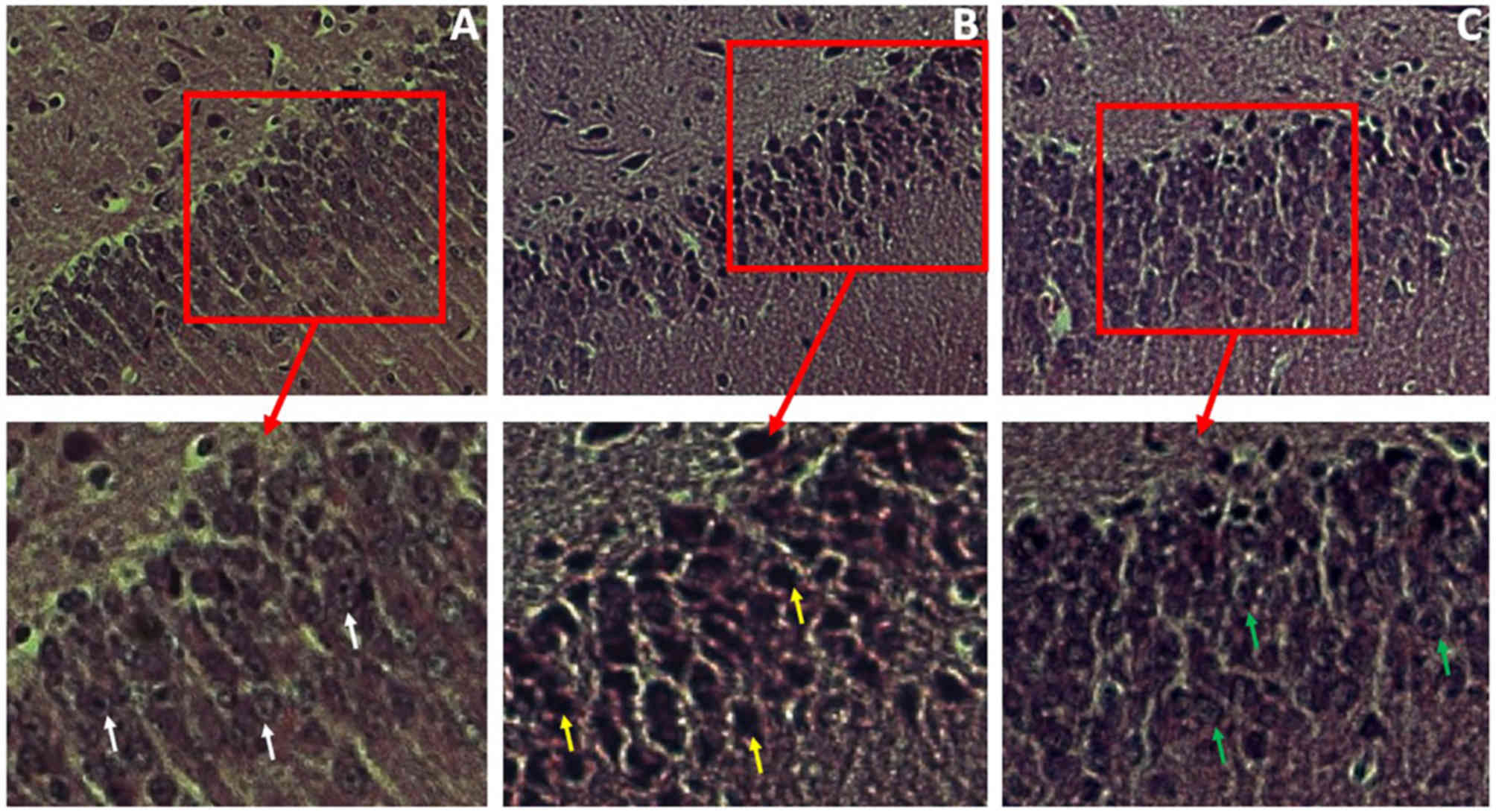
According to the TEM micrographs, the extent of
nuclear ultrastructural damage in the hippocampus was less
pronounced in the D-24 group compared with the N-24 group. The BC
group exhibited a nucleus that appeared normal with an intact
nuclear membrane (Fig. 8A), whereas
that in the N group was markedly damaged displaying chromatin
margination and distorted mitochondria (Fig. 8B). In the D group, these features of
damage were less visible (Fig. 8C).
In addition, autophagolysosomes (Fig.
8B; black arrows) and heteromorphic mitochondria (Fig. 8B; red arrows) could be observed in
the N group. However, little to none of these features were
identified in the D group.
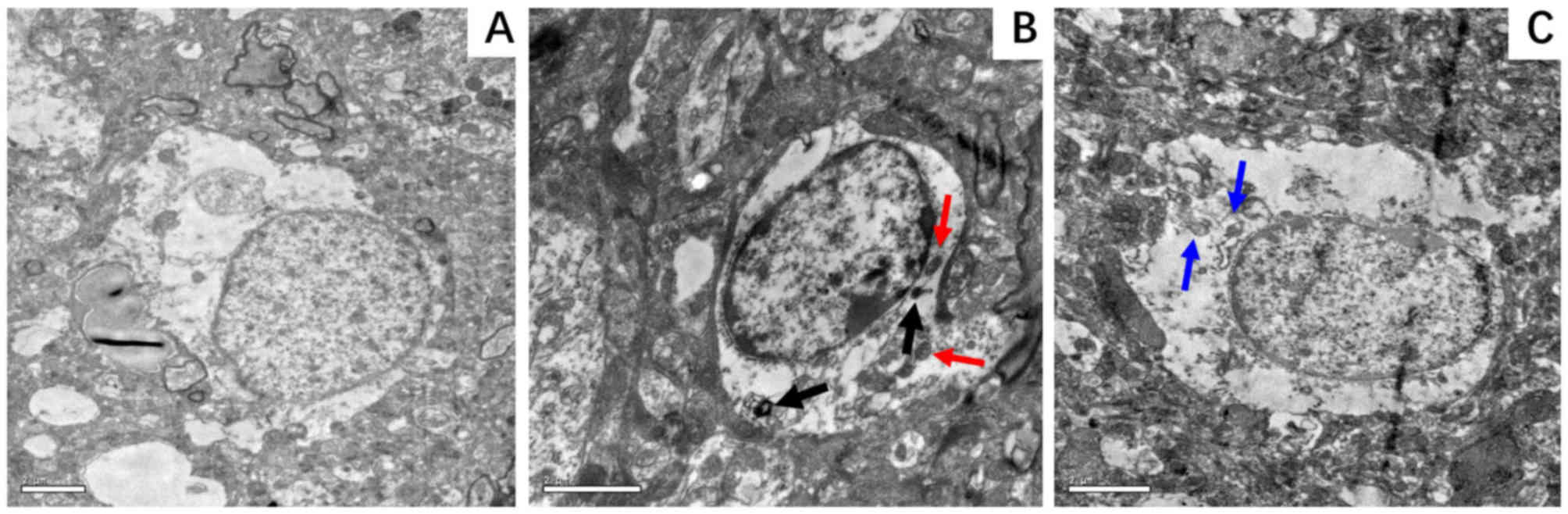 | Figure 8.Representative electron micrographs
of hippocampal neurocytes nuclei. Hippocampal tissue was isolated
from the rat hippocampus at 24 h following ROSC. (A) BC group,
demonstrating a normal nucleus with an intact nuclear membrane and
smooth chromatin; magnification, ×8,000. (B) N-24 group, displaying
a markedly damaged nucleus with chromatin margination and condensed
nucleoplasm, with autophagosomes (black arrows) and heteromorphic
mitochondria (red arrows) in the cytoplasm; magnification, ×12,000.
(C) D-24 group, displaying a slightly damaged nucleus with normal
nuclear membrane, slightly damaged chromatin and mitochondria (blue
arrows) in the cytoplasm; magnification, ×10,000. BC, blank control
group; D-24, dexmedetomidine group 24 h post-ROSC; N-24, normal
saline group 24 h post-ROSC; ROSC, return of spontaneous
circulation. |
Discussion
The present study found that Dex administration may
induce mild hypothermia, slow down heart rate, attenuate apoptosis
of neurocytes and improve neurological function after ROSC
following resuscitation from CA. This is supported by histological
and ultrastructural observations that Dex reduced damage in the rat
hippocampal tissue. Mechanistically, the protective effect of Dex
was found to be associated with the downregulation of pro-apoptotic
proteins Bax and Caspase-3 with a concomitant upregulation in
anti-apoptotic proteins Bcl-2 and HSP70. This is coupled with the
finding that the expression of autophagic protein Beclin1 was
downregulated in Dex-treated hippocampal tissue.
Dex is an α-2A adrenergic receptor agonist commonly
administered to patients as a sedative and analgesic (29). Previous studies have reported Dex to
be neuroprotective when combined with hypothermia (30,31).
However, few studies have assessed the association between Dex and
hypothermia, making the mechanism of this process unclear.
Lähdesmäki et al (32)
determined that the hypothermic effect of Dex may be abolished if
α-2A adrenergic receptor genes were knocked out in mice, which
indicated that the activation of α-2A adrenergic receptors, induced
by Dex, may inhibit neuronal firing and the release of monoamine
neurotransmitters associated with locomotor activity and body
temperature. The present study found that the core temperature and
heart rate in the group of rats treated with Dex was lower when
compared with the saline group in the same post-ROSC environment;
which corresponded well with the results from other studies. For
example, one previous report suggested that Dex can sustain
hypothermia by suppressing muscle shivering (33). It was hypothesized that this effect
is caused by the inhibition of neurotransmitter release from
sympathetic nerves located within the skeletal muscles by blocking
temperature-sensitive vanilloid-type transient receptor potential
ion channels (34). In addition, in
the sinoatrial node, Dex has been demonstrated to activate cardiac
Ca2+-sensitive potassium ion channels in pacemaker cells
during repolarization (35), causing
a reduction in heart rate.
It was demonstrated in our previous study that mild
hypothermia can reduce the level of lactic acid in the blood,
alleviate neurocyte apoptosis and improve neurological function in
rats that were resuscitated from CA (4). Sato et al (30) found in a rat model of focal cerebral
ischemia that short-term neurological outcomes could be improved by
a combination of Dex and hypothermia. In the current study, Dex
induced hypothermia and improved neurological function in rats
after ROSC following CA. However, no difference was found in lactic
acid levels between the two groups; this may be due to the
collection of arterial blood in 2 h intervals, which did not
provide enough time for lactic acid metabolism and excretion.
Another possibility was that the duration and severity of
hypothermia was not adequate for the elimination of lactic acid, or
that Dex may enhance the risk of metabolic acidosis (36).
Zhang et al (37) recently reported that HSP70 conferred
cellular protection against ischemic insults and possessed potent
anti-apoptotic properties in hippocampal neurons using a traumatic
brain injury rat model. To further understand the role of apoptosis
and autophagy in neurons after I/R, protein expression levels of
Bax, Bcl-2, Caspase-3 and Beclin1 were analyzed in the present
study. Bax, Caspase-3 and Beclin1 expression increased after ROSC,
suggesting that apoptosis and autophagy were activated by I/R in
the hippocampus. However, Dex administration inhibited this
increase, subsequently improving neurological function. Therefore,
it is possible that Dex may preserve neurological function by
reducing neural apoptosis and autophagy following I/R injury. A
number of previous studies have demonstrated that Dex can alleviate
I/R-induced neurocyte injury either by an intrinsic Bax/Caspase
pathway (38) or by inhibiting
neuronal autophagy through the upregulation of hypoxia-inducible
factor 1α expression (39). Bcl-2
binding to Beclin1 reduces its capacity to activate autophagy
without losing its own anti-apoptotic characteristics (40). The results of the present study
support the hypothesis that neuronal apoptosis and excessive
autophagy may be part of the pathophysiological process in I/R
injury following CA, a process that can at least in part be
alleviated by Dex.
The present study carries a number of limitations
that needs to be addressed in future investigations. The dosage and
method of Dex administration would need to be optimized and the
mechanism of Dex-induced hypothermia would require further
elucidation; both of which can be resolved by in vitro
studies. Furthermore, the background luminance of pathological
sections was inadequate, making images appear dark in color. In
addition, the present study was performed on a rat model in the
absence of underlying cardiac-cerebrovascular disease, hampering
applicability to human patients.
In the present study, it was found that I/R injury
activated Bax, Caspase-3 and Beclin1 expression, which indicated
that apoptosis and excessive autophagy may be the main pathological
process post-ROSC. The administration of Dex reduced Bax, Caspase-3
and Beclin1 expression levels, induced Bcl-2 and HSP70 expression
and hypothermia, and improved neurological function. Therefore, the
activation of intracellular apoptosis and autophagy following I/R
contributes to neural injury, whereas Dex treatment may induce
hypothermia, decrease cellular injury of neurons and improve
neurological function.
In conclusion, CA-induced I/R can lead to apoptosis
and excessive autophagy in hippocampal neurons. Dex treatment
induced hypothermia, in addition to exerting anti-apoptotic and
anti-autophagic effects to improve post-resuscitation neurological
function after CA. The present study uncovered a potential new
treatment strategy for CA recovery.
Acknowledgements
The authors would like to thank the Suzhou Health
Commission (Jiangsu, China) for their financial support, the
Experimental Center of the Second Affiliated Hospital of Soochow
University (Jiangsu, China) for their experimental guidance and the
Experimental Center of Suzhou Municipal Hospital and pathology
department of Suzhou Vocational Health College (Jiangsu, China) for
their technical assistance.
Funding
The current study was supported by a grant from the
Science and Technology Project ‘Health of Science and Education’ of
Suzhou Province (grant no. KJXW2013026).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JL proposed the current study and together with LJL,
drafted the manuscript. JL and ZL performed the experiments. ZL
collected and analyzed experimental data. SY and ZZ modified the
manuscript. ZC performed histological and ultrastructural analysis.
All authors read and approved the final manuscript for
publication.
Ethical approval and consent to
participate
The current study was performed according to the
exact guidelines and protocols for animal care and use established
by the Experimental Center of Suzhou Municipal Hospital (Animal
Experiment Permit Number, 120410), and was approved by the Suzhou
Municipal Hospital Ethic Committee (Ethical Review Number,
KL901008).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sanganalmath SK, Gopal P, Parker JR, Downs
RK, Parker JC Jr and Dawn B: Global cerebral ischemia due to
circulatory arrest: Insights into cellular pathophysiology and
diagnostic modalities. Mol Cell Biochem. 426:111–127. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Girotra S, Chan PS and Bradley SM:
Post-resuscitation care following out-of-hospital and in-hospital
cardiac arrest. Heart. 101:1943–1949. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ryu JA, Cho YH, Sung K, Choi SH and Yang
JH, Choi JH, Lee DS and Yang JH: Predictors of neurological
outcomes after successful extracorporeal cardiopulmonary
resuscitation. BMC Anesthesiol. 15:262015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu J, Qian HY, Liu LJ, Zhou BC, Xiao Y,
Mao JN, An GY, Rui MZ, Wang T and Zhu CL: Mild hypothermia
alleviates excessive autophagy and mitophagy in a rat model of
asphyxial cardiac arrest. Neurol Sci. 35:1691–1619. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu J, Shen Y, Liu LJ, Qian HY and Zhu CL:
Combining epinephrine and esmolol attenuates excessive autophagy
and mitophagy in rat cardiomyocytes after cardiac arrest. J
Cardiovasc Pharmacol. 66:449–456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu ZH, Cai M, Xiang J, Zhang ZN, Zhang JS,
Song XL, Zhang W, Bao J, Li WW and Cai DF: PI3K/Akt pathway
contributes to neuroprotective effect of Tongxinluo against focal
cerebral ischemia and reperfusion injury in rats. J Ethnopharmacol.
181:8–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin JY, Zhang MW, Wang JG, Li H, Wei HY,
Liu R, Dai G and Liao XX: Hydrogen sulfide improves neural function
in rats following cardiopulmonary resuscitation. Exp Ther Med.
11:577–587. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma Y, Chen C, Zhang S, Wang Q, Chen H,
Dong Y, Zhang Z, Li Y, Niu Z, Zhu T, et al: RNase alleviates
neurological dysfunction in mice undergoing cardiac arrest and
cardiopulmonary resuscitation. Oncotarget. 8:53084–53099.
2017.PubMed/NCBI
|
|
9
|
Zhang JC, Lu W, Xie XM, Pan H, Wu ZQ and
Yang GT: Mild hypothermia attenuates post-resuscitation brain
injury through a V-ATPase mechanism in a rat model of cardiac
arrest. Genet Mol Res. 15:2016.
|
|
10
|
Zhou T, Lin H, Jiang L, Yu T, Zeng C, Liu
J and Yang Z: Mild hypothermia protects hippocampal neurons from
oxygen-glucose deprivation injury through inhibiting caspase-3
activation. Cryobiology. 80:55–61. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rincon F: Targeted temperature management
in brain injured patients. Neurosurg Clin N Am. 29:231–253. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qin H, Tan W, Zhang Z, Bao L, Shen H, Wang
F, Xu F and Wang Z: 15d-prostaglandin J2 protects cortical neurons
against oxygen-glucose deprivation/reoxygenation injury:
Involvement of inhibiting autophagy through upregulation of Bcl-2.
Cell Mol Neurobiol. 35:303–312. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu X, Mao H, Liu J, Xu J, Cao J, Gu X and
Cui G: Dynamic change of SGK expression and its role in neuron
apoptosis after traumatic brain injury. Int J Clin Exp Pathol.
6:1282–1293. 2013.PubMed/NCBI
|
|
14
|
Zhou X, Liu Y, Huang Y, Zhu S, Zhu J and
Wang R: Hypertonic saline infusion suppresses apoptosis of
hippocampal cells in a rat model of cardiopulmonary resuscitation.
Sci Rep. 7:57832017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rizos IK, Tsoporis JN, Toumpoulis IK,
Salpeas V, Izhar S, Rigopoulos AG, Sakadakis EA and Parker TG:
Antiapoptotic effect of β1 blockers in ascending thoracic aortic
smooth muscle cells: The role of HSP70 expression. J Cardiovasc
Pharmacol. 72:86–96. 2018.PubMed/NCBI
|
|
16
|
Choudhary GS, Al-Harbi S and Almasan A:
Caspase-3 activation is a critical determinant of genotoxic
stress-induced apoptosis. Methods Mol Biol. 1219:1–9. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Y, Phuong Anh Nguyen T, Chen M and Xie
L: Tea polyphenols down-regulate JNK phosphorylation to inhibit
neuron apoptosis in rats with cardiac arrest. Zhonghua Wei Zhong
Bing Ji Jiu Yi Xue. 29:1122–1126. 2017.(In Chinese). PubMed/NCBI
|
|
18
|
Klionsky DJ and Ohsumi Y: Vacuolar import
of proteins and organelles from the cytoplasm. Annu Rev Cell Dev
Biol. 15:1–32. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi R, Weng J, Zhao L, Li XM, Gao TM and
Kong J: Excessive autophagy contributes to neuron death in cerebral
ischemia. CNS Neurosci Ther. 18:250–1260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu J, Shen Y, Qian HY, Liu LJ, Zhou BC,
Xiao Y, Mao JN, An GY, Rui MZ, Wang T and Zhu CL: Effects of mild
hypothermia on the ROS and expression of caspase-3 mRNA and LC3 of
hippocampus nerve cells in rats after cardiopulmonary
resuscitation. World J Emerg Med. 5:298–305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gu Y, Chen T, Li G, Xu C, Xu Z, Zhang J,
He K, Zheng L, Guan Z, Su X, et al: Lower Beclin 1 downregulates
HER2 expression to enhance tamoxifen sensitivity and predicts a
favorable outcome for ER positive breast cancer. Oncotarget.
8:52156–52177. 2016.PubMed/NCBI
|
|
22
|
Li L, You LS, Mao LP, Jin SH, Chen XH and
Qian WB: Combing oncolytic adenovirus expressing Beclin-1 with
chemotherapy agent doxorubicin synergistically enhances
cytotoxicity in human CML cells in vitro. Acta Pharmacol Sin.
39:251–260. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Afonso J and Reis F: Dexmedetomidine:
Current role in anesthesia and intensive care. Rev Bras Anestesiol.
62:118–133. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen X, Li L, Hu J, Zhang C, Pan Y, Tian D
and Tang Z: Anti-inflammatory effect of dexmedetomidine combined
with hypothermia on acute respiratory distress syndrome in rats. J
Surg Res. 216:179–184. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zakaria S, Kwong HJ, Sevransky JE,
Williams MS and Chandra-Strobos N: The cardiovascular implications
of sedatives in the cardiac intensive care unit. Eur Heart J Acute
Cardiovasc Care. 7:671–683. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yeda X, Shaoqing L, Yayi H, Bo Z, Huaxin
W, Hong C and Zhongyuan X: Dexmedetomidine protects against renal
ischemia and reperfusion injury by inhibiting the P38-MAPK/TXNIP
signaling activation in streptozotocin induced diabetic rats. Acta
Cir Bras. 32:429–439. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Geocadin RG, Ghodadra R, Kimura T, Lei H,
Sherman DL, Hanley DF and Thakor NV: A novel quantitative EEG
injury measure of global cerebral ischemia. Clin Neurophysiol.
111:1779–1787. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shelton KT, Qu J, Bilotta F, Brown EN,
Cudemus G, D'Alessandro DA, Deng H, DiBiasio A, Gitlin JA, Hahm EY,
et al: Minimizing ICU neurological dysfunction with
dexmedetomidine-induced sleep (MINDDS): Protocol for a randomised,
double-blind, parallel-arm, placebo-controlled trial. BMJ Open.
8:e0203162018.PubMed/NCBI
|
|
30
|
Sato K, Kimura T, Nishikawa T, Tobe Y and
Masaki Y: Neuroprotective effects of a combination of
dexmedetomidine and hypothermia after incomplete cerebral ischemia
in rats. Acta Anaesthesiol Scand. 54:377–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McAdams RM, McPherson RJ, Kapur R,
Phillips B, Shen DD and Juul SE: Dexmedetomidine reduces cranial
temperature in hypothermic neonatal rats. Pediatr Res. 77:772–778.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lähdesmäki J, Sallinen J, MacDonald E,
Sirviö J and Scheinin M: Alpha2-adrenergic drug effects on brain
monoamines, locomotion and body temperature are largely abolished
in mice lacking the alpha2A-adrenoceptor subtype.
Neuropharmacology. 44:882–892. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Callaway CW, Elmer J, Guyette FX,
Molyneaux BJ, Anderson KB, Empey PE, Gerstel SJ, Holquist K, Repine
MJ and Rittenberger JC: Dexmedetomidine reduces shivering during
mild hypothermia in waking subjects. PLoS One. 10:e01297092015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gifford JR, Ives SJ, Park SY, Andtbacka
RH, Hyngstrom JR, Mueller MT, Treiman GS, Ward C, Trinity JD and
Richardson RS: α1- and α2-adrenergic responsiveness in human
skeletal muscle feed arteries: The role of TRPV ion channels in
heat-induced sympatholysis. Am J Physiol Heart Circ Physiol.
307:H1288–H1297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Behmenburg F, Pickert E, Mathes A, Heinen
A, Hollmann MW, Huhn R and Berger MM: The cardioprotective effect
of dexmedetomidine in rats is dose-dependent and mediated by BKCa
channels. J Cardiovasc Pharmacol. 69:228–235. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma Y, Yu XY and Wang Y: Dose-related
effects of dexmedetomidine on immunomodulation and mortality to
septic shock in rats. World J Emerg Med. 9:56–63. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang MH, Zhou XM, Cui JZ, Wang KJ, Feng Y
and Zhang HA: Neuroprotective effects of dexmedetomidine on
traumatic brain injury: Involvement of neuronal apoptosis and HSP70
expression. Mol Med Rep. 17:8079–8086. 2018.PubMed/NCBI
|
|
38
|
Wu GJ, Chen JT, Tsai HC, Chen TL, Liu SH
and Chen RM: Protection of dexmedetomidine against
ischemia/reperfusion-induced apoptotic insults to neuronal cells
occurs via an intrinsic mitochondria-dependent pathway. J Cell
Biochem. 118:2635–2644. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Luo C, Ouyang MW, Fang YY, Li SJ, Zhou Q,
Fan J, Qin ZS and Tao T: Dexmedetomidine protects mouse brain from
ischemia-reperfusion injury via inhibiting neuronal autophagy
through up-regulating HIF-1α. Front Cell Neurosci. 11:1972017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ciechomska IA, Goemans GC, Skepper JN and
Tolkovsky AM: Bcl-2 complexed with Beclin-1 maintains full
anti-apoptotic function. Oncogene. 28:2128–2141. 2009. View Article : Google Scholar : PubMed/NCBI
|