Introduction
Metachromatic leukodystrophy (MLD; Mendelian of
Inheritance in Man #250100) is an autosomal recessive inherited
leukoencephalopathy characterized by demyelination of the central
and peripheral nervous systems (1).
These pathological changes are caused by failure of sulfatides and
other glycolipid-containing sulfuric acids to be desulfurized and
their subsequent deposition in the lysosomes of systemic tissues.
Desulfurization requires the combined efforts of the enzyme
arylsulfatase A (ASA; Enzyme Commission no. 3.1.6.8), encoded by
the ARSA gene, a lysosomal hydrolase, and sphingolipid activator
protein B, encoded by the PSAP gene. A deficiency in any of these
components results in abnormal metabolism of sulfatides, which
triggers progressive degenerative metabolic encephalopathy
(2,3). The worldwide prevalence of MLD is
estimated to be between 1 in 40,000 and 1 in 170,000 (4).
MLD is divided into three clinical subtypes
according to the age at onset of the disease: Late infantile (<4
years), juvenile (4–14 years) and adult (>14 years) (5). Clinical manifestations of MLD are
varied and lack specificity (6).
Three types of MLD are differentiated by the presence or absence of
neurological symptoms and the pattern of disease progression. The
severity of MLD appears to be negatively correlated with the age at
onset (7). Infantile MLD is the most
common form, accounting for ~60% of all diagnosed cases. The first
clinical symptoms of infantile MLD are mainly the deterioration of
the motor system, including spastic tetraparesis, frequent falls
and walking on toes. As the disease progresses, a patient may
develop flaccid paralysis and lose the ability to stand, exhibit
speech deficits and a decrease in psychological functions, develop
optic atrophy, suffer generalized or partial seizures and develop
peripheral neuropathy, causing death within 2–4 years from initial
diagnosis (8–10). The symptoms of juvenile MLD include
poor intellectual capabilities, emotional problems, language
disorders and a gradual regression in motor function. Patients with
juvenile MLD die within 10–15 years after the first symptoms
appear, with only few cases reaching their 20th birthday (11,12). The
adult form of the disease presents with neuromuscular or
behavioural problems and progresses slowly (12).
A genotype-phenotype correlation has been reported
in MLD (13–15). A deficiency in ASA is caused by
mutations in the ARSA gene. The ARSA gene (GenBank accession no.
NG_009260) is located on chromosome 22q13.33. This small gene (~3.5
kb) has eight exons and encodes 509 amino acid precursors (GenBank
accession nos. NM_000487.5 and NP_000478.3). Studies have indicated
that in all populations, the ARSA gene has three alleles (15–18). The
first allele is named ARSA-MLD (a pair of pathogenic alleles).
Polten et al (15) reported
that the ARSA-MLD gene has two different alleles. Mutations in one
allele, designated allele I (or O), result in non-functional
activity of ASA. Mutations in the other allele, designated allele A
(or R), maintain the residual activity of ASA. Patients who are
homozygous for allele I or allele A mainly present with the late
infantile form or the adult form of MLD, respectively. Patients who
are heterozygous for the two alleles (I/A) usually present with the
juvenile form (6). The second allele
is a pair of ASA pseudodeficiency (PD) alleles (ARSA-PD). Whether
the mutation is homozygous (ARSA-PD/ARSA-PD) or heterozygous
(ARSA-PD/ARSA-MLD) (17), the level
of ASA may be 5–15% of the reference value in certain patients with
the mutant ARSA-PD gene, while the structure of ASA is moderately
altered (19). In patients with a
low level of ASA, residual enzyme activity is sufficient for normal
physiological functions, which is known as PD. In European and
American populations, the most common ARSA-PD allele has two
variants (16). One is c.1049A>G
(N350S), which prevents the ASA protein from entering lysosomes.
The other is c.*96A>G, a mutation in the 3′ untranslated region
(poly A tail), which causes a significant reduction in ASA protein
production. However, Asian populations have only one allele
(c.1049A>G) of ARSA-PD. These Asian patients account for 20–30%
of all patients with MLD (20).
Furthermore, homozygosity for the mutant gene has little effect on
the activity of ASA. The third allele is (ARSA-MLD; ARSA-PD)
(18). It is reported that ARSA-MLD
and ARSA-PD, known as the ARSA-MLD-PD allele, are located on the
same chromosome. Based on the aforementioned genetic heterogeneity
of MLD, it is required to identify this multiple allele in the ARSA
gene to determine the genotype-phenotype correlation. To date, 254
mutations have been detected in the ARSA gene (21).
In the present study, whole-exome sequencing (WES)
was performed in the family of a case of MLD to identify causative
mutations and to offer a potential explanation for the
genotype-phenotype correlation in MLD.
Subjects and methods
Subjects and clinical evaluation
The patient of the present study was recruited at
Yuying Children's Hospital of Wenzhou Medical University (Wenzhou,
China). At the first medical examination of the male patient at the
age of 2 years 8 months due to a motor vehicle collision without
any obvious indication of resulting neurological damage, a brain
magnetic resonance imaging (MRI) scan was performed to ensure no
underlying brain injury. The imaging unexpectedly revealed signs of
MLD. Further enquiries regarding the proband's family history
unveiled that the proband's grandparents (maternal parents) had a
consanguineous marriage. Given the special family history,
cognitive assessments were performed using the Gesell Development
Scales test (22). It is the most
common test to assess neurobehavioural development in infants
(<3 years of age) through five factors (gross motor function,
fine motor function, speech, adaptive behaviour and interpersonal
behaviour). The results of Gesell Development Scales test was
determined as development quotient (DQ) (22). The patient was evaluated by
determining the DQ at the age of 2 years 8 months. Pediatric
subjects scoring DQ≤75, 75<DQ<85 and DQ≥85 exhibit low,
marginal and normal development, respectively (23). In addition, several complementary
inspections were performed to confirm the diagnosis, including ASA
activity in leukocytes, electromyography (EMG) and gas
chromatography-mass spectrometry (GC-MS) (24) at the proband's first visit. GC-MS
represents an unbiased and open approach that allows the detection
of unexpected changes in metabolite levels.
During 6 years of follow-up, the patient was
subjected to continuous physical assessments. The Wechsler
Intelligence Scale-third Edition (WAIS-III) test for
neurodevelopmental assessment (25,26) was
performed when the patient turned 4 years old. At our institution,
the WAIS-III test is commonly used to evaluate the
neurodevelopmental status of individuals aged ≥4 years (26). The results of the WAIS-III test are
expressed as an intelligence quotient (IQ), and the IQ is scored
according to the UK WAIS-III manual scoring criteria (25). An IQ of 90 is the boundary between
normal and low intelligence. The WAIS-III comprises 14 subtests
(vocabulary, similarities, information, comprehension, block
design, matrix reasoning, picture completion, picture arrangement,
coding, symbol search, digit span, letter-number sequencing,
arithmetics and object assembly) (27). The Ayres Sensory Integration (ASI)
test (28–30) was performed at the age of 7 years 11
months (July, 2017) to assess behavioural problems. Furthermore,
neuroradiological studies were performed by brain MRI scans as the
patient aged. At the most recent visit, the proband and the
proband's parents were subjected to genetic testing.
WES
Genomic DNA was isolated from peripheral blood
leukocytes of the affected proband and the proband's parents by
Kangso Medical Inspection. Whole-exome capture using the SureSelect
Human All Exon kit (Agilent Technologies, Inc.) and high-throughput
sequencing were performed in-house as previously described
(7,8). The reads were aligned for
single-nucleotide variant (SNV) calling and subsequent analysis for
prioritization of candidate genes (9). ANNOVAR (version 20180118) (31) was used to annotate the detected
variations. ANNOVAR can utilize annotation databases conforming to
Generic Feature Format version 3 (GFF3). The gene mutations we
verified based on the low frequency of the detected variants
provided in relevant gene databases [mainly the 1000 Genomes
Project (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/)
and dbSNP (https://www.ncbi.nlm.nih.gov/SNP/)]. First, DNA
samples were compared with the hg 19 reference (one version of
normal human genome from NCBI database). If the detected mutations
did not match the template file, a database was consulted (mainly
the 1000 Genomes Project). Subsequently, according to the frequency
of base mutations given in the database, it was determined whether
the mutation site is pathogenic. Furthermore, several studies
suggest that in numerous cases, variations in the chromosome copy
number (CNV), rearrangement and structure may be connected with
disease (32). Therefore, the CNV
was also determined by using CODEX (33) and XHMM software (34). CODEX and XHMM present a novel
normalization and CNV calling method. CODEX was used to remove
biases and artifacts in WES data, and produce accurate CNV calls.
The XHMM software was designed to recover information on CNVs from
targeted exome sequence data.
In silico analysis
Deleterious missense SNVs were predicted by the
following web-based tools: i) Sorting Intolerant From Tolerant
(SIFT; sift.bii.a-star.edu.sg/), where an
SNP with a SIFT score <0.05 predicts a negative effect on the
encoded amino acid, ii) polyphen-2 (genetics.bwh.harvard.edu/pph2/), where an SNP with a
score between 0.85 and 1.0 predicts a damaging effect on the
encoded amino acid, while an SNP with a score between 0.0 and 0.15
is predicted to be benign and an SNP with a score between 0.15 and
1.0 predicts a possibly damaging function, and iii) Mutation Taster
(www.mutationtaster.org/), where a score
close to 1 indicates a high ‘security’ of the prediction of the
given variant to be disease-causing.
In addition, multiple phylogenetic tree analyses of
ARSA homologs were performed using MEGA7 software (35).
Results
Clinical manifestation
A Chinese pedigree from the city of Wenzhou with an
index patient (MLD01) was recruited for the study (Fig. 1). The diagnosis of MLD was made
pre-symptomatically at the age of 2 years 8 months based on the
typical presentation of brain MRI. At first, the patient was
asymptomatic with generally normal development. Later, the proband
exhibited mild behavioural problems and was diagnosed with
attention deficit hyperactivity disorder (ADHD) at the age of 7
years 11 months based on the ASI examination. He had a normal gait
with no signs of paraparesis, spasticity or neuropathy, but was
socially immature based on his interactions with the surroundings
and individuals, as well as the ASI test. He was in a relatively
stable pre-onset stage at the time of writing. The result of the
EMG was normal. To date, the proband has developed no other
neurological symptoms and lives an ordinary life, except for
suffering from ADHD and struggling to keep up with his classmates
in a regular school.
ARSA activity determination
No biochemical abnormalities were detected in the
proband. ARSA activity in leukocytes of the proband was 150 nmol/17
h/mg protein at the first visit (at the age of 3 years in 2012) and
142 nmol/17 h/mg protein at the most recent visit (at the age of 8
years in 2017). Notably, the activity of ASA in 2017 was at the
lower end of the normal range (normal range: 134.1–325.1 nmol/17
h/mg protein).
Neurodevelopmental assessment
At the first visit at the age of 2 years 8 months in
March 2012, testing on the Gesell Development Scale revealed a
borderline DQ level (75.1). At 4 years of age (in 2013), WAIS-III
testing (36) suggested that the
patient had mild mental retardation (IQ, 81). Neurocognitive
evaluation in 2014 indicated a slight abnormality in full-scale IQ
with slow processing, low average working memory and below-average
executive skills. On the last follow-up (in 2017), the IQ had
reached an almost stable level at 80. In addition, ASI testing
suggested that the patient presented with inattention and bad
temper, and the patient was subsequently diagnosed with ADHD. Given
the mild symptoms, it was suggested that the patient could focus on
home education. The parents could have greater patience and provide
sufficient attention to the child. In addition, attention
exercises, including handwriting, were encouraged.
Neuroradiological studies
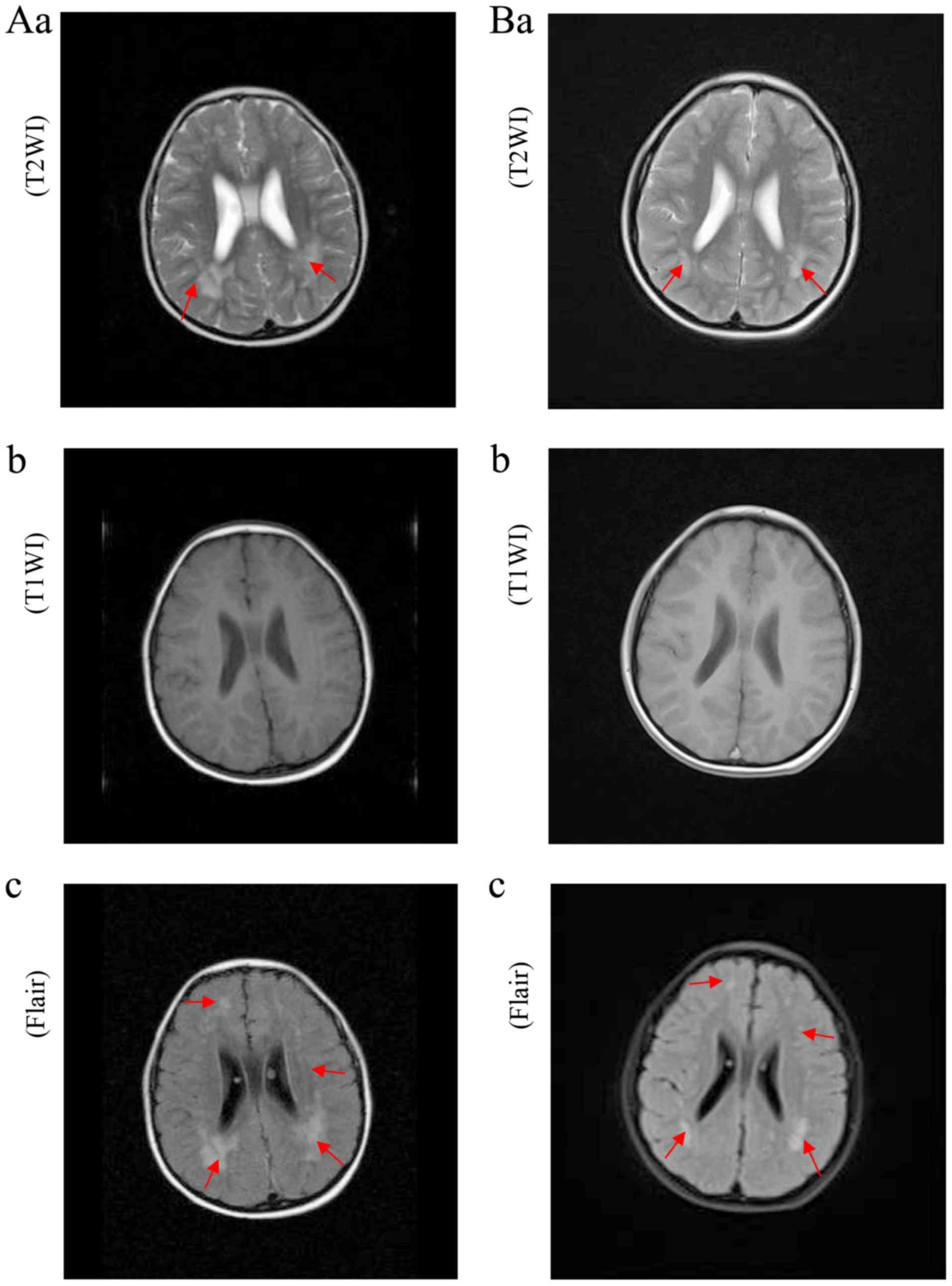
The images of the first MRI scan (Fig. 2Aa-c) in March 2012 exhibited a
lightly enlarged lateral ventricle and sporadic tigroid pattern of
white matter in the centrum semiovale and basal ganglia. These
results are characteristic of MLD. On the last MRI scan (Fig. 2Ba-c) in 2017, white-matter
hyperintensities were effectively unchanged. Over the 6-year
follow-up period, brain MRI scans revealed no evidence of
progressive demyelination, indicating that the patient is in a
stable condition.
Mutation detection
The mutation frequency may be expressed as that in
Asian populations (ASN) or in East Asians (EAS). The mutation
frequency of ‘c.1297C>G’ in the ARSA gene of the proband was
ASN=0.0035 and EAS=0.0020, respectively, and the mutation frequency
of ‘c.1345G>A’ in the ARSA gene of the proband was identical
(ASN=0.0035 and EAS=0.0020). Therefore, after variant filtering,
the study focused on the compound heterozygous variants
(c.1297C>G) + (c.1345G>A) in exon 8 in the ARSA gene of the
patient, which result in p.Leu433Val and p.Gly449Arg amino acid
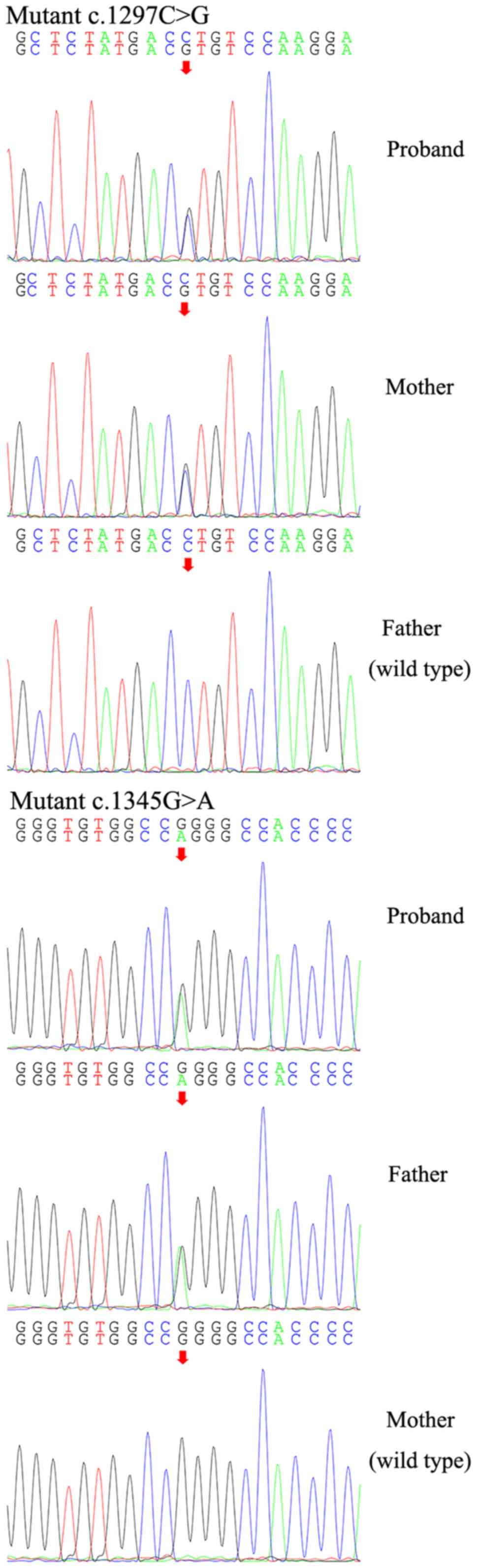
substitutions, respectively. Sanger sequencing was performed to
confirm the above results and to investigate the possible familial
segregation of the mutations. Compound heterozygous mutations
detected in the proband's parents indicated that each of the
proband's parents was a heterozygous carrier of either of the two
mutations (Fig. 3), who
coincidentally passed the mutation on to the proband individually.
Furthermore, no mutations with an autosomal-dominant pattern of
inheritance were identified. In addition, no suspicious CNVs were
identified in the WES data through the aforementioned computational
methods.
In silico analysis
To further characterize the missense mutations
identified in the family, different in silico tools were
used to assess the influences that the novel c.1297C>G and
c.1345G>A missense mutations have on ARSA protein function.
Pathogenicity predictions by the Mutation Taster programme
suggested that the c.1297C>G mutation is deleterious (Table I). Pathogenicity predictions by the
SIFT and PolyPhen-2 programme suggested that the c.1297C>G
mutation is tolerated and probably damaging, respectively. The
accuracy of Mutation Taster is reportedly the highest of the three
utilized tools (37).
 | Table I.Pathogenicity predictions for
p.Leu433Val and p.Gly449Arg mutations in ARSA by online in silico
prediction tools. |
Table I.
Pathogenicity predictions for
p.Leu433Val and p.Gly449Arg mutations in ARSA by online in silico
prediction tools.
| Amino acid
substitution | SIFTa |
PolyPhen-2b | Mutation
Tasterc |
|---|
| p.Leu433Val | Tolerated | Probably
damaging |
Disease-causing |
| p.Gly449Arg | Tolerated | Benign | – |
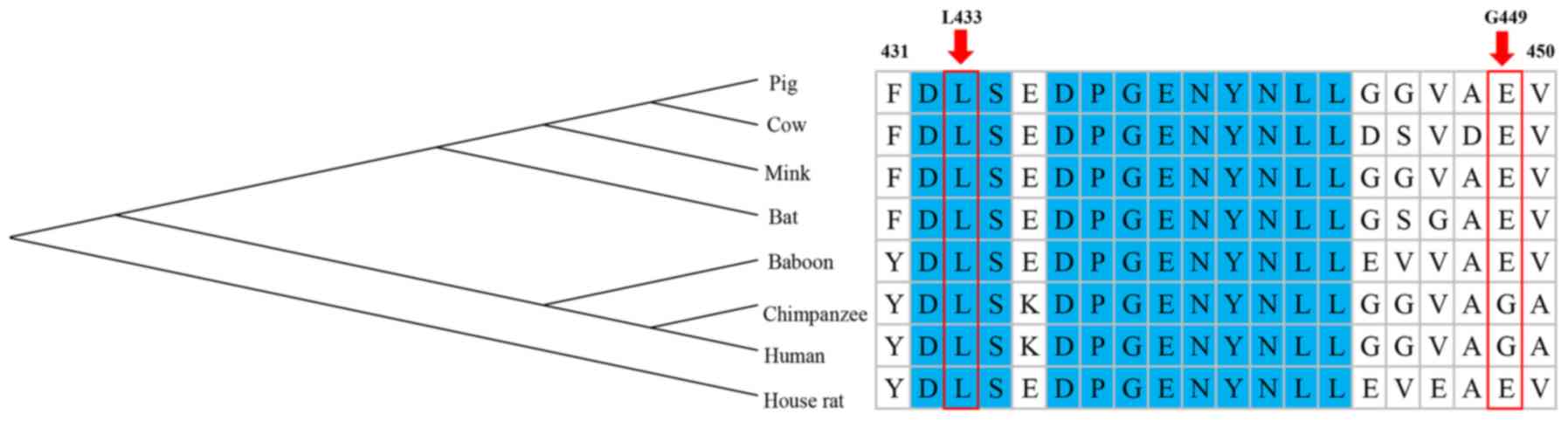
Furthermore, it is thought that highly conserved
amino acid sequences have a functional value and are significant
for protein structure, which indicates that they have an important
role in determining the conformation of different domains of a
protein (38). Multiple sequence
alignment of ARSA orthologues revealed that the amino acid L433 is
highly conserved across mammalian species, from human to rat, and
the amino acid G449 is less conserved. The evolutionary
conservation of the amino acid L433 indicates its significance in
the structure of the ASA protein (Fig.
4).
Discussion
The present study reported on a Chinese pediatric
patient diagnosed with MLD manifesting as ADHD as the major
clinical symptom. Of note, two missense mutations were identified
in the ARSA gene of the patient. The compound heterozygous variants
in the proband [(c.1297C>G) + (c.1345G>A)] were inherited
from the proband's mother and father, respectively. Of note, no
de novo variants were detected in the proband. According to
software prediction and comparison of homologous proteins, C.1297
was indicated to be the major mutation contributing to the onset of
the disease. By contrast, the patient's mother, who possesses the
same mutation, is normal. From the phenotype, the proband had a
tigroid pattern on every brain MRI and was diagnosed with mild
behavioural problems with MLD, whereas the proband's parents had no
symptoms. Notably, the subtype of adult MLD (>14 years) is
characterized by neuromuscular or behavioural problems. However,
the reported case showed behavioural problems at the age of 7
years. Therefore, it is likely that this presentation was due to
the compound heterozygous variants [(c.1297C>G) +
(c.1345G>A)].
Biochemical analysis indicated that the activity of
ASA was normal in the proband, which indicates that the mutations
in the two genes did not affect the enzyme level. Although test for
ASA activity in 2017 was at the lower end of the normal range, ASA
activity remained normal in patient's body. That is, there is no
qualitative change in ASA. Combined with the results of a previous
study, it may be hypothesized that c.1297C>G and c.1345G>A
probably belong to mutations in the ARSA-PD allele. It is reported
that the compound heterozygous mutation in ARSA-PD/ARSA-MLD is not
associated with progressive neurological disease (17).
Neurologically, all brain MRI scans for the proband
display pathological changes of demyelination, which is
characteristic of MLD. Furthermore, the patient's MRI images
exhibit little progression during the 6-year follow-up period. It
may be hypothesized that this may be associated with the normal
enzyme levels in the proband.
Concerning the phenotype, the patient's condition
cannot be regarded as severe when compared to other cases of MLD.
Given that the disease is usually classified according to the age
of onset, it is difficult to classify this case into a proper
subtype of MLD. The patient presented with ADHD-like symptoms but
had no neurological signs at the age of 7 years 11 months. The MRI
images exhibited demyelination of the cerebrum at the age of 2
years 8 months. If this case was to be classified according to the
age of onset, the patient should be diagnosed with juvenile MLD,
according to the clinical symptoms, the patient, who displayed
behavioural problems, should be diagnosed with the adult form.
Therefore, subjects with suspicion of MLD should be comprehensively
evaluated. The clinical manifestation of MLD is highly variable. If
clinicians diagnosed MLD only based on the general indicators of
the three subtypes, numerous patients would be misdiagnosed or the
diagnosis would be missed. Subjects with suspected MLD,
particularly in a consanguineous family, may be diagnosed through
genetic testing. In additionally, the phenotype is the result of
interactions between genes and the environment. A tigroid pattern
of white matter at the level of the centrum semiovale and basal
ganglia hyperintensity indicated in MRI highly suggests a diagnosis
of MLD, especially when considering the consanguinity relationship
between the child's grandparents. The clinical presentation of the
proband of the present study is atypical, which may be associated
with his living environment. Of note, he possessed a large number
of good educational resources and his parents provided a nurturing
environment and sufficient attention.
However, the present study only included one family.
It may be worthwhile to perform a cohort study to further examine
genotype-phenotype associations in MLD. In addition, experimental
animal studies with the same mutations are required to investigate
the biochemical characterization of MLD (39). By enlarging the number of samples for
mutation analysis, it is possible to screen those subjects at risk
or with symptoms to diagnose early. The authors of the current
study may be able to summarise some common features by increasing
the size of the cohort in future studies. As physician can
recognize people at high risk of MLD, only individual gene test
need to be performed rather than WES. This is helpful for detecting
ARSA gene mutations quickly and reliably, and reducing the cost of
genetic testing.
In conclusion, the compound heterozygous variants
(c.1297C>G) + (c.1345G>A) were described in a Chinese
pediatric patient with MLD. The patient, having normal activity of
ASA, presented with ADHD-like symptoms, no neurological symptoms
and obvious signs of MLD on MRI.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
XC and YYW contributed the central idea and analyzed
most of the data. YYW wrote the initial draft of the paper. CL,
SMW, QFX, QH, SC and YWL contributed to refining the ideas,
carrying out additional analyses and finalizing this paper.
Ethical approval and consent to
participate
Informed consent was obtained from the proband's
parents. The present study was approved by the ethics committee of
Wenzhou Medical University (Wenzhou, China).
Patient consent for publication
The patient's parents were contacted by telephone to
obtain verbal informed consent.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MLD
|
metachromatic leukodystrophy
|
|
ASA
|
arylsulfatase A
|
|
WES
|
whole-exome sequencing
|
|
ADHD
|
attention deficit hyperactivity
disorder
|
|
MRI
|
magnetic resonance imaging
|
|
DQ
|
development quotient
|
|
IQ
|
intelligence quotient
|
|
EMG
|
electromyography
|
|
WAIS-III
|
Wechsler intelligence scale-third
edition
|
|
SNV
|
single-nucleotide variant
|
|
T1WI
|
T1-weighted imaging
|
|
CNV
|
copy number variation
|
References
|
1
|
Coulter-Mackie MB, Gagnier L, Beis MJ,
Applegarth DA, Cole DEC, Gordon K and Ludman MD: Metachromatic
leucodystrophy in three families from Nova Scotia, Canada: a
recurring mutation in the arylsulphatase A gene. Journal of Medical
Genetics. 34:493–498. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aggarwal S, Yurlova L and Simons M:
Central nervous system myelin: Structure, synthesis and assembly.
Trends Cell Biol. 21:585–593. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bosio A, Binczek E and Stoffel W:
Functional breakdown of the lipid bilayer of the myelin membrane in
central and peripheral nervous system by disrupted
galactocerebrosides synthesis. Proc Natl Acad Sci USA.
93:13280–13285. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brimley CJ, Lopez J, van Haren K, Wilkes
J, Sheng X, Nelson C, Korgenski EK, Srivastava R and Bonkowsky JL:
National variation in costs and mortality for leukodystrophy
patients in US children's hospitals. Pediatr Neurol. 49:156–162,e1.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shahzad MA, Khaliq S, Amar A and Mahmood
S: Metachromatic leukodystrophy (MLD): A pakistani family with
novel ARSA gene mutation. J Mol Neurosci. 63:84–90. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gieselmann V, Polten A, Kreysing J and von
Figura K: Molecular genetics of metachromatic leukodystrophy. J
Inherit Metab Dis. 13:222–227. 1994.
|
|
7
|
Simell O: The metabolic and molecular
bases of inherited disease. JAMA. 286:23292001.
|
|
8
|
Lugowska A, Mierzewska H,
Bekiesińska-Figatowska M, Szczepanik E, Goszczańska-Ciuchta A and
Bednarska-Makaruk M: A homozygote for the c.459+1G>A mutation in
the ARSA gene presents with cerebellar ataxia as the only first
clinical sign of metachromatic leukodystrophy. J Neurol Sci.
338:214–217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barboura I, Ferchichi S, Dandana A,
Jaidane Z, Ben Khelifa S, Chahed H, Ben Mansour R, Chebel S, Maire
I and Miled A: Metachromatic leucodystrophy. Clinical, biological,
and therapeutic aspects. Ann Biol Clin (Paris). 68:385–391.
2010.(In French). PubMed/NCBI
|
|
10
|
Kehrer C, Groeschel S, Kustermann-Kuhn B,
Bürger F, Köhler W, Kohlschütter A, Bley A, Steinfeld R, Gieselmann
V and Krägeloh-Mann I; German LEUKONET, : Language and cognition in
children with metachromatic leukodystrophy: Onset and natural
course in a nationwide cohort. Orphanet J Rare Dis. 9:182014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barrell C: Juvenile metachromatic
leukodystrophy: Understanding the disease and implications for
nursing care. J Pediatr Oncol Nurs. 24:64–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Patil SA and Maegawa GH: Developing
therapeutic approaches for metachromatic leukodystrophy. Drug Des
Devel Ther. 7:729–745. 2013.PubMed/NCBI
|
|
13
|
Gieselmann V, Fluharty AL, Tønnesen T and
Von Figura K: Mutations in the arylsulfatase A pseudodeficiency
allele causing metachromatic leukodystrophy. Am J Hum Genet.
49:407–413. 1991.PubMed/NCBI
|
|
14
|
Regis S, Corsolini F, Stroppiano M, Cusano
R and Filocamo M: Contribution of arylsulfatase A mutations located
on the same allele to enzyme activity reduction and metachromatic
leukodystrophy severity. Hum Genet. 110:351–355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Polten A, Fluharty AL, Fluharty CB,
Kappler J, von Figura K and Gieselmann V: Molecular basis of
different forms of metachromatic leukodystrophy. N Engl J Med.
324:18–22. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tinsa F, Caillaud C, Vanier MT, Bousnina
D, Boussetta K and Bousnina S: An unusual homozygous arylsulfatase:
A pseudodeficiency in a metachromatic leukodystrophy tunisian
patient. J Child Neurol. 25:82–86. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Penzien JM, Kappler J, Herschkowitz N,
Schuknecht B, Leinekugel P, Propping P, Tønnesen T, Lou H, Moser H,
Zierz S, et al: Compound heterozygosity for metachromatic
leukodystrophy and arylsulfatase A pseudodeficiency alleles is not
associated with progressive neurological disease. Am J Hum Genet.
52:557–564. 1993.PubMed/NCBI
|
|
18
|
Fluharty AL: Arylsulfatase A
Deficiency-GeneReviews®-NCBI Bookshelf. University of
Washington Seattle. 2014.
|
|
19
|
Aubourg P, Sevin C and Cartier N: Mouse
Models of Metachromatic Leukodystrophy and Adrenoleukodystrophy. De
Deyn P and Van Dam D: Animal Models of Dementia. Neuromethods,
Humana Press. 48:493–513. 2011. View Article : Google Scholar
|
|
20
|
Shi H: Metachromatic Leukodystrophy.
Chinese Journal of Practical Pediatrics. 507–510. 2009.(In
Chinese).
|
|
21
|
The Human Gene Mutation Database at the
Institute of Medical Genetics in Cardiff. http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ARSAApril
23–2019
|
|
22
|
Fishler K, Graliker BV and Koch R: The
predictability of intelligence with gesell developmental scales in
mentally retarded infants and young children. Am J Ment Defic.
69:515–525. 1965.PubMed/NCBI
|
|
23
|
Huang XN, Zhang Y, Feng WW, Wang HS, Cao
B, Zhang B, Yang YF, Wang HM, Zheng Y, Jin XM, et al: Reliability
and validity of warning signs checklist for screening
psychological, behavioral and developmental problems of children.
Zhonghua Er Ke Za Zhi. 55:445–450. 2017.(In Chinese). PubMed/NCBI
|
|
24
|
Roessner U, Wagner C, Kopka J, Trethewey
RN and Willmitzer L: Technical advance: Simultaneous analysis of
metabolites in potato tuber by gas chromatography-mass
spectrometry. Plant J. 23:131–142. 2010. View Article : Google Scholar
|
|
25
|
Dumont R and Willis JO: Wechsler Adult
Intelligence Scale-3rd edition. John Wiley. (Sons, Inc). 2008.
|
|
26
|
Wechsler D: Wechsler Adult Intelligence
Scale-3rd edition. 1997.
|
|
27
|
Cockcroft K, Alloway T, Copello E and
Milligan R: A cross-cultural comparison between South African and
British students on the Wechsler Adult Intelligence Scales Third
Edition (WAIS-III). Front Psychol. 6:2972015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mailloux Z, Mulligan S, Roley SS, Blanche
E, Cermak S, Coleman GG, Bodison S and Lane CJ: Verification and
clarification of patterns of sensory integrative dysfunction. Am J
Occup Ther. 65:143–151. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mulligan S: Patterns of sensory
integration dysfunction: A confirmatory factor analysis. Am J
Occupational Therapy. 52:819–828. 1998. View Article : Google Scholar
|
|
30
|
Parham LD, Roley SS, May-Benson TA, Koomar
J, Brett-Green B, Burke JP, Cohn ES, Mailloux Z, Miller LJ and
Schaaf RC: Development of a Fidelity Measure for Research on the
Effectiveness of the Ayres Sensory Integration(R) Intervention[J].
American Journal of Occupational Therapy. 65:133–142. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang K, Li M and Hakonarson H: Hakonarson,
ANNOVAR: Functional annotation of genetic variants from
high-throughput sequencing data. Nucleic Acids Res. 38:e1642010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Carter NP: Methods and strategies for
analyzing copy number variation using DNA microarrays. Nat Genet 39
(7 Suppl). S16–S21. 2007. View
Article : Google Scholar
|
|
33
|
Jiang Y, Oldridge DA, Diskin SJ and Zhang
NR: CODEX: A normalization and copy number variation detection
method for whole exome sequencing. Nucleic Acids Res. 43:e392015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fromer M and Purcell SM: Purcell, using
XHMM software to detect copy number variation in whole-exome
sequencing data. Curr Protoc Hum Genet. 81:7.23.1–21. 2014.
View Article : Google Scholar
|
|
35
|
Tamura K, Peterson D, Peterson N, Stecher
G, Nei M and Kumar S: MEGA5: Molecular evolutionary genetics
analysis using maximum likelihood, evolutionary distance, and
maximum parsimony methods. Mol Biol Evol. 28:2731–2739. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Iverson GL: Interpreting change on the
WAIS-III/WMS-III in clinical samples. Arch Clin Neuropsychol.
16:183–191. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shahzad MA, Khaliq S, Amar A and Mahmood
S: Metachromatic leukodystrophy (MLD): A pakistani family with
novel ARSA gene mutation. J Mol Neurosci. 63:84–90. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Özkan A and Özkara HA: Metachromatic
leukodystrophy: Biochemical characterization of two
(p.307Glu->Lys, p.318Trp->Cys) arylsulfatase A mutations.
Intractable Rare Dis Res. 5:280–283. 2016. View Article : Google Scholar : PubMed/NCBI
|