Introduction
Diabetic cardiomyopathy (DCM), a heart disease that
is independent of hypertension and coronary artery disease, mainly
manifests as a cardiac dysfunction and a structural disorder. DCM
increases the risk of heart failure and is one of the major causes
of death in patients with diabetes (1). Pathogenesis of DCM is multifactorial,
but hyperglycemia-induced overproduction of superoxides by the
mitochondrial electron-transport chain is considered to be the
common pathway leading to the condition (2). Mitochondria participate in the terminal
part of the majority of apoptotic signaling pathways as
‘gatekeepers’ that ultimately determine cell survival and death
(3). Mitochondrial dysfunction is
closely related to multiple morbidities associated with DCM. A
previous study demonstrated that compared with the control, the
mass-specific respiration of streptozotocin-diabetic hearts was
reduced during oxidative phosphorylation (4). Mitochondria in atrial tissues in
individuals with type 2 diabetes demonstrated a markedly reduced
capacity for glutamate and fatty acid-supported respiration
compared with those of non-diabetic patients (5).
Tumor necrosis factor receptor-associated protein 1
(TRAP1) is a heat shock protein (HSP), which belongs to the HSP90
family, mainly located in mitochondria. TRAP1 is involved in a
series of cell signaling pathways, participating in the regulation
of mitochondrial function and cell oxidative stress (6). TRAP1 can protect cells and reduce
apoptosis by decreasing the production of reactive oxygen species
(ROS) and reducing oxidative stress (6). TRAP1 protects cancer cells from
hypoxia-induced mitochondrial dysfunction and cell apoptosis during
tumor progression, and is a key regulator of mitochondrial
bioenergy that assists tumor cells in avoiding death and damage
(7). Therefore, TRAP1 is a potential
therapeutic target for designing novel anticancer agents (8). Previous studies determined that changes
in TRAP1 expression in normal tissues also impact cellular
function. TRAP1 serves a role in protecting mitochondria against
damaging stimuli via the decrease of ROS generation (9). TRAP1 reduces cardiac hypertrophy and
fibrosis that is induced by pressure overload through blocking the
TAK/P38, JNK, and AKT signaling pathways (10). TRAP1 overexpression decreases ROS
production and preserves mitochondrial membrane potential during
glucose deprivation, while preserving ATP levels and cell viability
during oxygen-glucose deprivation (11). However, the role of TRAP1 in DCM and
its mechanisms of action remain undefined.
The present study aimed to investigate the role of
TRAP1 in hyperglycemia-induced myocardial damage and to understand
the associated mechanisms in cardiomyocytes under high glucose
conditions.
Materials and methods
H9c2 cell culture
H9c2 cells (The Cell Bank of Type Collection of
Chinese Academy of Sciences) were cultured with low-glucose DMEM
medium (Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.), 1% penicillin-streptomycin
solution (Gibco; Thermo Fisher Scientific, Inc.) and incubated with
5% CO2 at 37°C. When cell density reached 70–80%
confluence, the cells were passaged in a 1:3 ratio, and logarithmic
growth phase cells were used in subsequent experiments. The cells
were grouped into normal glucose control group (C; 5.5 mmol/l
glucose treatment for 48 h; Sigma-Aldrich; Merck KGaA), high
glucose group (G; 33 mmol/l glucose treatment for 48 h), negative
control small interfering RNA group (NC siRNA; negative control
siRNA was transfected into H9c2 cells for 24 h, then cultured in
the aforementioned hyperglycemic condition for 48 h) and TRAP1
siRNA group (TRAP1 siRNA was transfected into H9c2 cells for 24 h,
then cultured in the aforementioned hyperglycemic conditions for 48
h).
siRNA design and preparation
TRAP1 siRNA and NC siRNA sequences were designed and
synthesized by GenePharma Co., Ltd. The sequences of the siRNAs
were as follows: TRAP1 siRNA forward, 5′-GCUAGUGGAUCAGAUCUAUTT-3′
and reverse, 5′-AUAGAUCUGAUCCACUAGCTT-3′; NC siRNA forward,
5′-UUCUCCGAACGUGUCACGUTT-3′, and reverse:
5′-ACGUGACACGUUCGGAGAATT-3′. The siRNA products were vacuum
centrifuged (2,000 × g at room temperature for 5 min) and dissolved
in diethyl pyrocarbonate-treated water (GenePharma Co., Ltd.) to
obtain a 20 µM stock solution before use.
siRNA transfection
H9c2 cells were seeded in six-well plates
(1.5×105 cells/well) and cultured for 12 h to obtain
fully adherent cells with a confluence of 30–50%. Transfection was
performed by adding 13 µl of 20 µM TRAP1 siRNA stock or NC siRNA
stock and 6 µl Lipofectamine® RNAiMAX (Invitrogen;
Thermo Fisher Scientific, Inc.) to 400 µl Opti-MEM medium (Gibco;
Thermo Fisher Scientific, Inc.). Following gentle mixing, the
mixture was incubated at room temperature for 5 min to form a
siRNA-transfection reagent mixture, which was then added to the
six-well plate containing the cells, followed by addition of
FBS-free DMEM to a final volume of 2 ml/well. The medium was
replaced with complete DMEM medium following 6 h. After
transfection with siRNA for 24 h, the medium was replaced with high
glucose DMEM for 48 h prior to subsequent experimentation.
Western blot analysis
Cells were washed twice with PBS, followed by
addition of radioimmunoprecipitation assay lysis buffer (Beyotime
Institute of Biotechnology) for 10 min. Next, the samples were kept
on ice, sonicated (130 W; 20 kHz; 30%; 4°C; 10 sec), centrifuged
(13,680 × g; 4°C; 5 min) and the supernatant was added to the
loading buffer (Hangzhou Fude Biological Technology Co., Ltd.) and
boiled for 10 min. Protein concentration was determined using a BCA
assay. The proteins (30 µg per lane) were separated via SDS-PAGE on
a 10% gel then transferred to polyvinylidene fluoride membranes.
Membranes were blocked using 5% non-fat dry milk at room
temperature for 2 h. Samples were incubated with primary antibodies
anti-TRAP1 (1:1,000; cat. no. ab176478; Abcam) and GAPDH (1:3,000;
cat. no. 5174; Cell Signaling Technology, Inc.) overnight at 4°C.
Following washes, membranes were incubated with horseradish
peroxidase-conjugated goat anti-rabbit secondary antibody (1:5,000;
cat. no. L153B; GeneCopoeia, Inc.) for 1 h at room temperature.
This was followed by use of the FDbio-Femto Enhanced
chemiluminescence kit (Hangzhou Fude Biological Technology Co.,
Ltd.) and exposure of the plate to a chemiluminescence imaging
analyzer. Image J software (Image J 64-bit Java 1.8.0_112; National
Institutes of Health) was used to analyze the gray-scale value of
each band with GAPDH used as a loading control.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using TRIzolÒ
(Thermo Fisher Scientific, Inc.). The quality and quantity of RNA
samples were determined using Epoch™ microplate spectrophotometer
(BioTek Instruments, Inc.). Total RNA was reverse transcribed into
cDNA according to the instructions of PrimeScriptTM RT master mix
(Takara Bio, Inc). qPCR was performed using Applied Biosystems™
PowerUp™ SYBR™ Green mix (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. The reaction
volume total was 30 µl and PCR was performed with
LightCycler® 480. the 2−ΔΔCq method was used
for quantification (12). The
primers used were as follows: TRAP1 forward,
5′-CTCAGTTGCTACAGCCCACA-3′ and reverse, 5′-CTGCTATCATGGCGTTCTCA-3′;
GAPDH forward, 5′-AGGTGAAGGTCGGAGTCAAC-3′; and reverse,
5′-CGCTCCTGGAAGATGGTGAT-3′.
MTS assay to determine cell
viability
H9c2 cells were plated in 96-well plates at 5,000
cells/well. Viability was measured using the CellTiter
96® AQueous One Solution Assay kit (Promega
Corporation). Culture medium was replaced with complete medium (100
µl/well) at the respective treatment time points. Then 20 µl MTS
assay reagent per well was added and incubated for 2 h at 37°C. A
microplate reader was used to detect the optical density (OD) at
490 nm. The optical density is directly proportional to cell
viability.
Intracellular ROS detection
ROS were detected using
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) probe
(Beyotime Institute of Biotechnology) according to the
manufacturer's guidelines and fluorescence intensity was observed
under a fluorescence microscope (magnification, ×40). Fluorescence
intensity was proportional to intracellular ROS levels. Image J
software (Image J 64-bit Java 1.8.0_112; National Institutes of
Health) was used to analyze the staining intensity of each
group.
Mitochondrial membrane potential
JC-1 fluorescence staining mitochondrial membrane
potential detection kit (Beyotime Institute of Biotechnology) was
used to detect changes in mitochondrial membrane potential
according to the manufacturer's instructions. Fluorescence
intensity was observed under a fluorescence microscope
(magnification, 40×). Image J software (Image J 64-bit Java
1.8.0_112; National Institutes of Health) was used to analyze the
staining intensity value of each group.
Statistical analysis
All data were analyzed using SPSS 16.0 statistical
software (SPSS, Inc.) and expressed as the mean ± standard
deviation. This analysis was performed for three independent
experiments at least. The differences between groups were analyzed
using one-way analysis of variance followed by Least Significance
Difference post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
TRAP1 inhibition significantly
decreases TRAP1 protein and gene expression
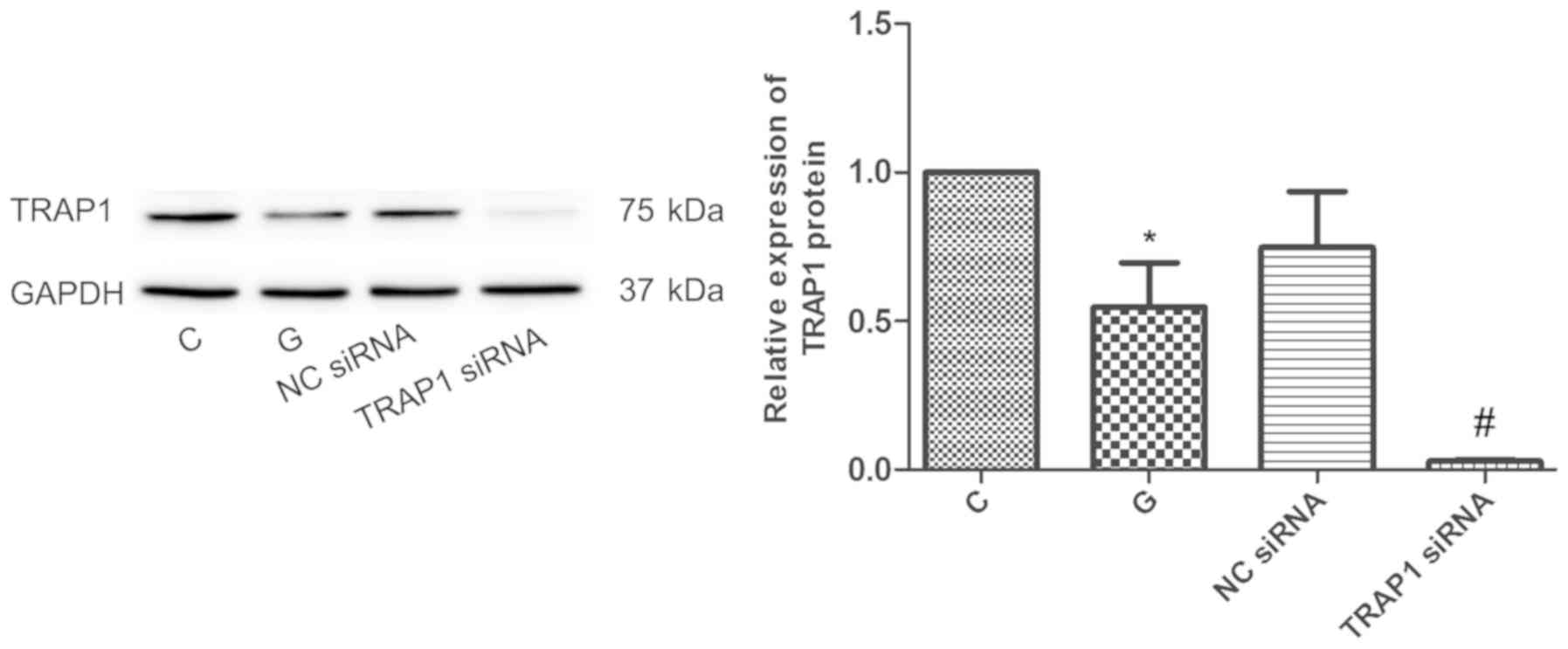
siRNA-mediated knockdown efficiency was evaluated by
detecting TRAP1 protein and mRNA levels by western blot analysis
and RT-qPCR, respectively. Compared with C group, the expression of
TRAP1 protein was decreased significantly by 45% in G group
(P<0.05; Fig. 1). Compared with
the NC siRNA group, the expression of TRAP1 was significantly
decreased by 96% in TRAP1 siRNA group (P<0.05; Fig. 1). Compared with C group, the level of
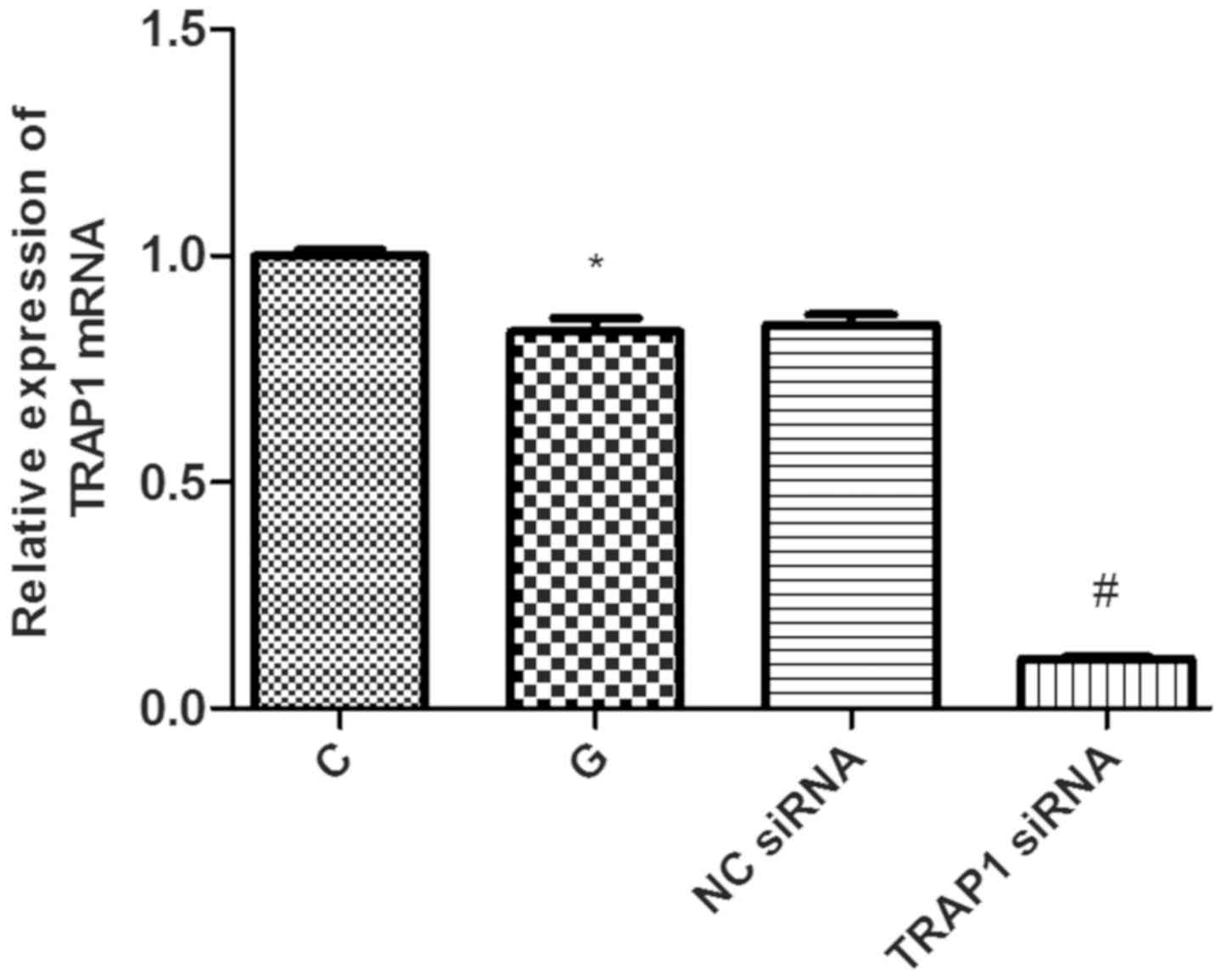
TRAP1 mRNA was decreased significantly by 13% in G group
(P<0.05; Fig. 2). Compared with
NC siRNA group, the level of TRAP1 mRNA was significantly decreased
by 87% in TRAP1 siRNA group (P<0.05; Fig. 2).
Downregulation of TRAP1 reduces cell
viability
Compared with the NC siRNA group, the downregulation
of TRAP1 significantly reduced cell viability of H9c2 cells
(P<0.05; Fig. 3). Relative cell
viability values in the normal glucose control group, high glucose
group, NC siRNA group and TRAP1 siRNA group were 1.02±0.07,
0.83±0.06, 0.83±0.07 and 0.65±0.05, respectively. The optical
density is directly proportional to cell viability. The viability
of G group was significantly lower compared with C group
(P<0.05; Fig. 3). Downregulation
of TRAP1 significantly reduced cell viability of H9c2 cells
compared with the NC siRNA group (P<0.05; Fig. 3).
Downregulation of TRAP1 leads to ROS
overproduction
The level of intracellular ROS can reflect the
degree of oxidative stress in cells. Excessive ROS production can
induce mitochondrial dysfunction of cardiomyocytes, thereby causing
apoptosis (13). ROS content was
investigated to determine whether cardiomyocyte injury was caused
by the downregulation of TRAP1. The results demonstrated that the
green fluorescence intensity of the G group was higher compared
with C group (P<0.05; Fig. 4).
The green fluorescence intensity is directly proportional to
intracellular ROS content, and therefore, these results indicate
that H9c2 cells produced more ROS in the hyperglycemic state.
Compared with NC siRNA group, the green fluorescence of TRAP1 siRNA
group was significantly enhanced, suggesting that downregulation of
TRAP1 in H9c2 cells led to ROS overproduction (P<0.05; Fig. 4).
Downregulation of TRAP1 lowers the
mitochondrial membrane potential
To investigate the effect of TRAP1 on mitochondrial
function, the mitochondrial membrane potential in H9c2 cells was
investigated (Fig. 5). Results
demonstrated that the red fluorescence intensity of G group was
weaker compared with C group (P<0.05; Fig. 5), suggesting that mitochondrial
membrane potential of H9c2 cells was reduced when cells were in the
hyperglycemic state. Red fluorescence of cardiomyocytes transfected
with TRAP1 siRNA was weaker (P<0.05; Fig. 5) compared with NC siRNA group,
indicating that downregulation of TRAP1 induced mitochondrial
dysfunction in cardiomyocytes.
Discussion
DCM is one of the major causes of death in patients
with diabetes (14). Insulin
resistance, hyperinsulinemia and hyperglycemia are important
factors triggering the development of DCM (15). The mechanisms involve multiple
pathophysiological changes, including metabolic derangements,
inappropriate activation of the renin-angiotensin-aldosterone
system, oxidative stress, inflammation and immune dysfunction
(16). Oxidative stress is
considered to be an initiation factor for DCM development.
Hyperglycemia and glucose fluctuation can induce acute oxidative
stress, which leads to ROS accumulation, cellular dysfunction and
cell death (17). The present study
demonstrated that ROS levels increased and cardiomyocyte viability
decreased when cells were in the hyperglycemic state, which was
consistent with the literature (18,19).
TRAP1 was first identified as a partner of tumor
necrosis factor receptor 1 (TNFR1) in a yeast two-hybrid screen
(20). Subsequently, it was
demonstrated to be a molecular chaperone of the HSP90 family, as
TRAP1 and HSP90 display 34% identity and 60% homology at the mRNA
level and share the same domain organization (20). Previous studies demonstrated a
pivotal role of TRAP1 in the pathogenesis of different tumors
including breast, colon, pancreas, lung and nasopharyngeal
adenocarcinoma with TRAP1 significantly upregulated in these
malignancies (21–23). TRAP1 inhibition leads to impaired
cell proliferation, mitochondrial lesion and reduced energy
metabolism, and eventually results in apoptosis of tumor cells
(24).
TRAP1 is variably expressed in multiple organs and
tissues, including skeletal muscle, liver, heart, brain, kidney,
pancreas, lung and placenta (20).
It is mainly localized in the inner membrane of the mitochondria,
and its primary function is to protect cells from mitochondrial
apoptosis (6). TRAP1 is part of a
supramolecular complex that is physically associated with HSP90,
HSP60 and cyclophilin D (CypD) (25). CypD is a matrix peptidyl prolyl
isomerase, which is a component of the organellar mitochondrial
permeability transition pore (mPTP). This complex regulates
mitochondrial function by controlling the mitochondrial
permeability transition pore, cytochrome C release and the caspase
cascade (26). TRAP1 can inhibit the
opening of mPTP and reduce the release of cytochrome C and caspase
3 (27). Sorcin, a calcium binding
protein of 18 kDa, is a TRAP1 client protein. Overexpression or
downregulation of sorcin and/or TRAP1 demonstrated a reciprocal
regulation between these two proteins and revealed that their
interaction is required for sorcin mitochondrial localization and
TRAP1 stability (28). Sorcin has a
key role in controlling mitochondrial Ca2+ homeostasis,
which assists in regulating the opening of the mPTP. TRAP1
chaperone activity may be crucial for sorcin folding, which
contributes to mitochondrial Ca2+ homeostasis and is
involved in Ca2+-dependent mPTP regulation (6). Silencing of TRAP1 via RNA interference
increases ROS accumulation, whereas TRAP1 overexpression attenuates
ROS production (29). Although TRAP1
is considered as ‘mitochondrial HSP90’, it also exists in
extramitochondrial locations. TRAP1 interacts with the proteasomal
particle TBP7 on the outer side of the endoplasmic reticulum (ER),
and serves an important role in protein quality control. Inhibition
of TRAP1 or TBP7 increases ER stress, apoptosis and intracellular
protein ubiquitination (30,31). Previous studies demonstrated that
hypoxia induces an increase in TRAP1 expression in cardiomyocytes,
and TRAP1 has a protective role by regulating the opening of the
mPTP (32–34). TRAP1 provides cardioprotection
against myocardial ischemia reperfusion by ameliorating
mitochondrial dysfunction (35).
The present study demonstrated that TRAP1 levels
were reduced in cardiomyocytes under hyperglycemic conditions,
which was accompanied by increased ROS production, decreased
mitochondrial membrane potential and impaired cell viability.
Silencing of TRAP1 led to further deterioration of these changes,
which indicated that TRAP1 may be involved in oxidative stress,
mitochondrial dysfunction and cardiomyocyte lesion during
hyperglycemia. However, the present study only involves preliminary
in vitro research. Further in vitro and in
vivo studies are required to investigate the role and
underlying mechanisms of TRAP1 in DCM and to reinforce the current
findings. Future work will involve establishment of a model of
TRAP1 overexpression to investigate the changes of cardiomyocytes
under high glucose surroundings.
In conclusion, the present study demonstrated that
TRAP1 was associated with oxidative stress and mitochondrial
dysfunction in cardiomyocytes cultured under hyperglycemic
conditions. This suggested that TRAP1 may be involved in DCM
development. To the best of our knowledge, this is the first study
to investigate the expression and role of TRAP1 in cells that were
in a high glucose state.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Natural Science Foundation of Guangdong, China (grant no.
2017A030310257), Science and Technology Program of Guangzhou, China
(grant no. 201707010045) and the Doctoral Start-up Fund of
Guangzhou Medical University (grant no. 2015C10).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ and WL designed experiments. ZZ and XZ performed
experiments and analyzed data. ZZ drafted this manuscript and all
authors approved this manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dei Cas A, Fonarow GC, Gheorghiade M and
Butler J: Concomitant diabetes mellitus and heart failure. Curr
Probl Cardiol. 40:7–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Granville DJ and Gottlieb RA:
Mitochondria: Regulators of cell death and survival.
ScientificWorldJournal. 2:1569–1578. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pham T, Loiselle D, Power A and Hickey AJ:
Mitochondrial inefficiencies and anoxic ATP hydrolysis capacities
in diabetic rat heart. Am J Physiol Cell Physiol. 307:C499–C507.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anderson EJ, Kypson AP, Rodriguez E,
Anderson CA, Lehr EJ and Neufer PD: Substrate-specific derangements
in mitochondrial metabolism and redox balance in the atrium of the
type 2 diabetic human heart. J Am Coll Cardiol. 54:1891–1898. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matassa DS, Amoroso MR, Maddalena F,
Landriscina M and Esposito F: New insights into TRAP1 pathway. Am J
Cancer Res. 2:235–248. 2012.PubMed/NCBI
|
|
7
|
Masgras I, Sanchez-Martin C, Colombo G and
Rasola A: The chaperone TRAP1 as a modulator of the mitochondrial
adaptations in cancer cells. Front Oncol. 7:582017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lettini G, Maddalena F, Sisinni L,
Condelli V, Matassa DS, Costi MP, Simoni D, Esposito F and
Landriscina M: TRAP1: A viable therapeutic target for future cancer
treatments? Expert Opin Ther Targets. 21:805–815. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Im CN, Lee JS, Zheng Y and Seo JS: Iron
chelation study in a normal human hepatocyte cell line suggests
that tumor necrosis factor receptor-associated protein 1 (TRAP1)
regulates production of reactive oxygen species. J Cell Biochem.
100:474–486. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Y, Jiang DS, Yan L, Cheng KJ, Bian
ZY and Lin GS: HSP75 protects against cardiac hypertrophy and
fibrosis. J Cell Biochem. 112:1787–1794. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Voloboueva LA, Duan M, Ouyang Y, Emery JF,
Stoy C and Giffard RG: Overexpression of mitochondrial Hsp70/Hsp75
protects astrocytes against ischemic injury in vitro. J Cereb Blood
Flow Metab. 28:1009–1016. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Roul D and Recchia FA: Metabolic
alterations induce oxidative stress in diabetic and failing hearts:
Different pathways, same outcome. Antioxid Redox Signal.
22:1502–1514. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bragg F, Holmes MV, Iona A, Guo Y, Du H,
Chen Y, Bian Z, Yang L, Herrington W, Bennett D, et al: Association
between diabetes and cause-specific mortality in rural and urban
areas of China. JAMA. 317:280–289. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee WS and Kim J: Diabetic cardiomyopathy:
Where we are and where we are going. Korean J Intern Med.
32:404–421. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jia G, Whaley-Connell A and Sowers JR:
Diabetic cardiomyopathy: A hyperglycaemia- and
insulin-resistance-induced heart disease. Diabetologia. 61:21–28.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu ZW, Zhu HT, Chen KL, Dong X, Wei J,
Qiu C and Xue JH: Protein kinase RNA-like endoplasmic reticulum
kinase (PERK) signaling pathway plays a major role in reactive
oxygen species (ROS)-mediated endoplasmic reticulum stress-induced
apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol.
12:1582013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huynh K, Bernardo BC, Mcmullen JR and
Ritchie RH: Diabetic cardiomyopathy: Mechanisms and new treatment
strategies targeting antioxidant signaling pathways. Pharmacol
Ther. 142:375–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Q, Wang S and Cai L: Diabetic
cardiomyopathy and its mechanisms: Role of oxidative stress and
damage. J Diabetes Investig. 5:623–634. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song HY, Dunbar JD, Zhang YX, Guo D and
Donner DB: Identification of a protein with homology to hsp90 that
binds the type 1 tumor necrosis factor receptor. J Biol Chem.
270:3574–3581. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang B, Wang J, Huang Z, Wei P, Liu Y,
Hao J, Zhao L, Zhang F, Tu Y and Wei T: Aberrantly upregulated
TRAP1 is required for tumorigenesis of breast cancer. Oncotarget.
6:44495–44508. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gao JY, Song BR, Peng JJ and Lu YM:
Correlation between mitochondrial TRAP-1 expression and lymph node
metastasis in colorectal cancer. World J Gastroenterol.
18:5965–5971. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee JH, Kang KW, Kim JE, Hwang SW, Park
JH, Kim SH, Ji JH, Kim TG, Nam HY, Roh MS, et al: Differential
expression of heat shock protein 90 isoforms in small cell lung
cancer. Int J Clin Exp Pathol. 8:9487–9493. 2015.PubMed/NCBI
|
|
24
|
Yuan M and Li X: Research progress of the
relationship between TRAP1 and malignant tumor. Shi Yong Zhong Liu
Xue Za Zhi. 31:371–375. 2017.(In Chinese).
|
|
25
|
Montesano Gesualdi N, Chirico G, Pirozzi
G, Costantino E, Landriscina M and Esposito F: Tumor necrosis
factor-associated protein 1 (TRAP-1) protects cells from oxidative
stress and apoptosis. Stress. 10:342–350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kang BH, Plescia J, Dohi T, Rosa J, Doxsey
SJ and Altieri DC: Regulation of tumor cell mitochondrial
homeostasis by an organelle-specific Hsp90 chaperone network. Cell.
131:257–270. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Altieri DC, Stein GS, Lian JB and Languino
LR: TRAP-1, the mitochondrial Hsp90. Biochim Biophys Acta 1823.
767–773. 2012.
|
|
28
|
Landriscina M, Laudiero G, Maddalena F,
Amoroso MR, Piscazzi A, Cozzolino F, Monti M, Garbi C, Fersini A,
Pucci P and Esposito F: Mitochondrial chaperone Trap1 and the
calcium binding protein Sorcin interact and protect cells against
apoptosis induced by antiblastic agents. Cancer Res. 70:6577–6586.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hua G, Zhang Q and Fan Z: Heat shock
protein 75 (TRAP1) antagonizes reactive oxygen species generation
and protects cells from granzyme M-mediated apoptosis. J Biol Chem.
282:20553–20560. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Amoroso MR, Matassa DS, Laudiero G,
Egorova AV, Polishchuk RS, Maddalena F, Piscazzi A, Paladino S,
Sarnataro D, Garbi C, et al: TRAP1 and the proteasome regulatory
particle TBP7/Rpt3 interact in the endoplasmic reticulum and
control cellular ubiquitination of specific mitochondrial proteins.
Cell Death Differ. 19:592–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Amoroso MR, Matassa DS, Sisinni L, Lettini
G, Landriscina M and Esposito F: TRAP1 revisited: Novel
localizations and functions of a ‘next-generation’ biomarker
(review). Int J Oncol. 45:969–977. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xiang F, Huang YS, Shi XH and Zhang Q:
Mitochondrial chaperone tumour necrosis factor receptor-associated
protein 1 protects cardiomyocytes from hypoxic injury by regulating
mitochondrial permeability transition pore opening. FEBS J.
277:1929–1938. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xiang F, Ma SY, Lv YL, Zhang DX, Song HP
and Huang YS: Tumor necrosis factor receptor-associated protein 1
regulates hypoxia-induced apoptosis through a
mitochondria-dependent pathway mediated by cytochrome c oxidase
subunit II. Burns Trauma. 7:162019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiang F, Zhang DX, Ma SY and Huang YS:
Mechanism of protective effects of tumor necrosis factor receptor
associated protein 1 on hypoxic cardiomyocytes of rats. Zhonghua
Shao Shang Za Zhi. 32:744–751. 2016.(In Chinese). PubMed/NCBI
|
|
35
|
Zhang P, Lu Y, Yu D, Zhang D and Hu W:
TRAP1 provides protection against myocardial ischemia-reperfusion
injury by ameliorating mitochondrial dysfunction. Cell Physiol
Biochem. 36:2072–2082. 2015. View Article : Google Scholar : PubMed/NCBI
|