Introduction
Heart failure (HF) is the leading cause of death
worldwide (1). Hypertension the most
important independent risk factor for HF, which may result in
pathological left ventricular (LV) hypertrophy and myocardial
fibrosis (2–5). Previous studies have revealed that
inhibiting or reversing cardiac hypertrophy may be an effective
treatment for preventing LV remodeling and HF (6–8).
Therefore, there is a requirement for the identification of novel
drugs that improve pathological myocardial remodeling in patients
with HF.
Schisandrin B (Sch B) is the most abundant and
potent ingredient isolated from the fruit of Schisandra spp.
and is a popular Chinese herbal medicine (9). Sch B exhibits a number of
pharmacological effects, including anti-inflammatory, antioxidative
and anticancer effects (10–12). A previous study indicated that Sch B
exhibits anti-inflammatory activity via modulation of the
redox-sensitive transcription factors Nrf2 and NF-κB (11). A study performed by Ip et al
(10) demonstrated that Sch B
protects against carbon tetrachloride toxicity through enhanced the
function of the hepatic glutathione antioxidant system. A number of
studies have indicated that Sch B serves a vital role in
cardiovascular disease. A report by Thandavarayan et al
(13) has demonstrated that Sch B
prevents doxorubicin induced cardiac dysfunction by modulation of
DNA damage, oxidative stress and inflammation through inhibition of
MAPK/p53 signaling. Furthermore, Chen et al (14) observed that Sch B reduces
inflammation, inhibits apoptosis, and improves cardiac function
following myocardial infarction. A number of studies have
consistently demonstrated that Sch B ameliorates myocardial
ischemia-reperfusion injury (15,16).
However, the mechanism of action of Sch B in stress-induced
pathological cardiac hypertrophy has not been investigated. The aim
of the present study was therefore to investigate the protective
effect of Sch B on stress-induced pathological cardiac hypertrophy
and to elucidate its underlying mechanism.
Materials and methods
Experimental animals
All in vivo experiments were approved by the
Animal Care and Use Committee of the Central Hospital of Wuhan. The
36 C57BL/6 mice (male; age, 7–8 weeks; weight, 22–26 g) were
purchased from Beijing HFK Bioscience Co., Ltd. Mice were housed in
an environment with controlled light cycles (12 h light/dark),
temperature (20–24°C) and humidity (45–55%). Food and water were
provided ad libitum.
Mouse model and experimental
grouping
Surgeries and subsequent analyses were performed in
a blinded manner. Aortic banding (n=24) and sham (n=12) surgeries
were performed as previously described (17). Briefly, 36 mice were anesthetized
with an intraperitoneal injection of 3% (50 mg/kg) sodium
pentobarbital solution. The depth of anesthesia during the surgical
procedures was assessed via the pedal withdrawal reflex and
breathing rate. An incision was made on the left side of the chest
of each mouse and the thoracic aorta was identified by blunt
dissection at the second intercostal space. A 26-G needle was
placed on the thoracic aorta and a 7-0 silk suture was placed
around the thoracic aorta and needle, and tied. The needle was
withdrawn immediately after successful ligation, resulting in a
transverse aortic constriction (TAC). Doppler analysis was
subsequently performed to ensure that the aorta had been adequately
constricted. The thoracic aortas of the animals in the sham group
were threaded but not ligated.
Sch B treatment protocol in vivo
Sch B (purity ≥97%) was purchased from Shanghai
PureOne Biotechnology Co., Ltd. and dissolved in olive oil. Oral
administration is currently the main route of administration of Sch
B in clinical practice (9).
Therefore, in order to better simulate clinical treatment, the mice
in the current study were administered Sch B intragastrically.
Furthermore, intragastric administration of Sch B has previously
been reported by Chen et al (14). A study on the pharmacokinetics of Sch
B revealed that the calculated absolute oral bioavailability of Sch
B was ~55.0% for female rats and 19.3% for male rat (18). A number of studies also support the
good oral bioavailability of Sch B in mice (19,20).
Therefore, SchB was not decomposed or destroyed by gastric acid.
Following ligation of the thoracic aorta, animals in the Sch B
group (n=12) received 80 mg/kg Sch B intragastrically per day for 4
weeks. Animals in the TAC (n=12) and sham (n=12) groups received
the same volume of olive oil daily.
Echocardiography analysis
The mice were anesthetized with 1.5–2% isoflurane
and echocardiography using a Mylab30CV (Esaote Group) ultrasound
system with a 15-Mz probe was performed, 4 weeks after the TAC. The
short-axis view of the standard left ventricular papillary muscle
was selected, and the left ventricular end-systolic diameter
(LVESd), left ventricular end-diastolic diameter (LVEDd), left
ventricular ejection fraction (LVEF) and left ventricular
fractional shortening (LVFS) were measured. Following
echocardiography, the mice were sacrificed via an overdose of
sodium pentobarbital (200 mg/kg) injected intraperitoneally. The
body weight (BW), heart weight (HW), lung weight (LW) and tibia
length (TL) were measured. HW/BW, LW/BW and HW/TL values were
subsequently calculated.
Myocardial histopathology
The heart was removed from the sacrificed animals
and placed in 10% potassium chloride to extrude the blood from the
heart cavity. Tissues were subsequently fixed in 4%
paraformaldehyde at 4°C for 12 h, dehydrated with a descending
alcohol series (100% alcohol for 5 min, 95% alcohol for 5 min and
75% alcohol for 5 min), embedded in paraffin and sectioned.
Cross-sections of the LV papillary muscle were prepared, and 4–5
mm-thick heart sections were subjected to conventional
hematoxylin-eosin (H&E; hematoxylin for 10 min and eosin for 5
min at room temperature) and Masson staining (alkali fuchsine
solution for 3 min, phosphomolybdic acid for 1 min, and aniline
blue solution for 2 min at room temperature). Sections were
observed under a microscope and Image Pro Plus software (version
6.0; Media Cybernetics, Inc.) was used to calculate the myocardial
cell cross-sectional area (CSA) and the area of the fibrotic region
in the LV.
Cell culture and angiontensin II (Ang
II) treatment
The rat cardiomyocyte cell line H9c2 was purchased
from the The Cell Bank of Type Culture Collection of the Chinese
Academy of Sciences and cultured in DMEM (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.), 2 mmol/l glutamine, 1 mmol/l pyruvate, 100 U/ml
penicillin and 100 mg/ml streptomycin. Cells were maintained in a
humidified incubator at 37°C and 5% CO2.
Previous studies indicated that pretreatment with
Sch B at concentration of 5–20 µM for 16–24 h resulted in optimal
myocardial protection against ischemic injury in vitro
(14,21). In the current study, cells were
pretreated with Sch B (5, 10 and 20 µM) for 16 h at room
temperature. The cells were subsequently treated with Ang II
dissolved in DMSO to a final concentration of 1 µM for 30 min at
room temperature. DMSO was used as the vehicle control. Cell
viability was measured using the MTT assay, which relies on the
ability of living cells to convert thiazole blue into purple
formazan. The optical density of the formazan was subsequently
detected at a wavelength of 570 nm. The results were normalized to
those of the untreated control. Protein and mRNA expression levels
in the treated and control cells were analyzed by western blot
analysis and reverse-transcription quantitative PCR (RT-qPCR),
respectively.
Western blot analysis
Total proteins were extracted from LV tissues and
H9c2 cells using lysis buffer containing Protease Inhibitor
Cocktail (cat. no. 04693159001; Roche Diagnostics),
Phenylmethanesulfonyl Fluoride (cat. no. AS1006; Aspen) and
Phosphatase Inhibitor (cat. no. AS1008; Aspen). Extracted proteins
were subjected to centrifugation (13,000 × g; 4°C; 5 min),
sonication (20 kHz; room temperature; 10 sec; 3 times) and heat
denaturation. The total protein concentration was determined using
the BCA Protein Assay kit (cat. no. P0010; Beyotime Institute of
Biotechnology). Proteins (40 µg/lane) were separated on an 8–10%
SDS-PAGE gel and transferred onto a polyvinylidene fluoride
membrane, which was subsequently incubated with primary antibodies
overnight at 4°C. Following primary antibody incubation, the
membrane was incubated with GAPDH (1:10,000; cat. no. ab37168;
Abcam) for 2 h at room temperature. The antibodies used in the
current study are presented in Table
I. Protein bands were detected using chemiluminescence (cat.
no. NCI 5079; Thermo Fisher Scientific, Inc.). Gray-scale scanning
and quantification were performed using Image J software (version
1.32; National Institutes of Health).
 | Table I.Mouse primary antibodies for western
blotting. |
Table I.
Mouse primary antibodies for western
blotting.
| Antibody | Source
organism | Supplier | Dilution | Cat. no. |
|---|
| ANP | Rabbit | Abcam | 1:1,000 | ab225844 |
| BNP | Rabbit | Abcam | 1:1,000 | ab236101 |
| β-MHC | Mouse | Abcam | 1:500 | ab11083 |
| Collagen I | Rabbit | Abcam | 1:500 | ab34710 |
| CTGF | Rabbit | Abcam | 1:1,000 | ab6992 |
| Collagen III | Rabbit | Abcam | 1:1,000 | ab7778 |
| P-ERK1/2 | Rabbit | Cell Signaling
Technology, Inc. | 1:1,000 | #4370 |
| ERK1/2 | Rabbit | Cell Signaling
Technology, Inc. | 1:2,000 | #4695 |
| P-JNK1/2 | Rabbit | Cell Signaling
Technology, Inc. | 1:1,000 | #4671 |
| JNK1/2 | Rabbit | Cell Signaling
Technology, Inc. | 1:1,000 | #9252 |
| P-P38 | Rabbit | Cell Signaling
Technology, Inc. | 1:1,000 | #4511 |
| P38 | Rabbit | Cell Signaling
Technology, Inc. | 1:2,000 | ab170099 |
| GAPDH | Rabbit | Abcam | 1:10,000 | ab37168 |
RT-qPCR
Total RNA was extracted from H9c2 cells using
TRIzol® reagent (cat. no. 15596-026; Thermo Fisher
Scientific, Inc.) and was reversed transcribed into cDNA using the
PrimeScript RT reagent kit (cat. no. RR047A; Takara Biotechnology
Co., Ltd.). The temperature protocol was 25°C for 10 min, 50°C for
60 min, 90°C for 5 min, and 4°C for 10 min. RT-qPCR was
subsequently performed using a 20 µl reaction system containing
cDNA, forward and reverse primers and SYBR® Premix Ex
Taq (cat. no. RR420A; Takara Biotechnology Co., Ltd.). The initial
denaturation was 95°C for 30 sec, 40 cycles of denaturation at 95°C
for 5 sec, annealing at 60°C for 30 sec and elongation at 72°C for
30 sec, and a final elongation at 72°C for 10 min. GAPDH was used
as an internal reference gene and presented as an expression fold
change using the 2−ΔΔCq method (22). The primers were synthesized by
Shanghai GenePharma Co., Ltd. The sequences of the primers used for
RT-qPCR are presented in Table
II.
| Table II.Mouse primers for reverse
transcription-quantitative PCR. |
Table II.
Mouse primers for reverse
transcription-quantitative PCR.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| Collagen I |
AACAAGGGAGGAGAGAGTGC |
AGTCTCTTGCTTCCTCCCAC |
| Collagen III |
GAAGGGCAGGGAACAACTGA |
GGGCAGTCTAGTGGCTCATC |
| CTGF |
CTGTGGGAGAAAACACCCCA |
CACTCTTCCAGGAGGCTCAC |
| ANP |
ATTGACAGGATTGGAGCCCA |
CAGAGTGGGAGAGGTAAGGC |
| BNP |
AGTCCTAGCCAGTCTCCAGA |
AGTCCTAGCCAGTCTCCAGA |
| β-MHC |
GGAGGAGATCAGTGAGAGGC |
GCTTCACCCGCTGTAGATTG |
| GAPDH |
TGAAGGGTGGAGCCAAAAG |
AGTCTTCTGGGTGGCAGTGAT |
Statistical analysis
SPSS statistical software (version 21; IBM Corp.)
was used to conduct all the data analysis. The results are
expressed as the mean ± SEM. Each experiment was repeated four
times. Statistical comparisons among multiple groups were performed
with one-way ANOVA followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Cardiac function and structure
No deaths were observed in the mice during the
4-week experimental period. Fig. 1A
shows the representative mouse M-mode echocardiograms in each
group. The LVEDd and LVESd were significantly lower in the Sch
B-treated group compared with the TAC group (P<0.05; Fig. 1B and C). The LVEF and LVFS were
significantly higher in the Sch B-treated group compared with the
TAC group (P<0.05; Fig. 1D and
E). In addition, the HW/BW, HW/TL and LW/BW values of the Sch
B-treated group were significantly lower compared with the TAC
group (P<0.05; Fig. 1F-H).
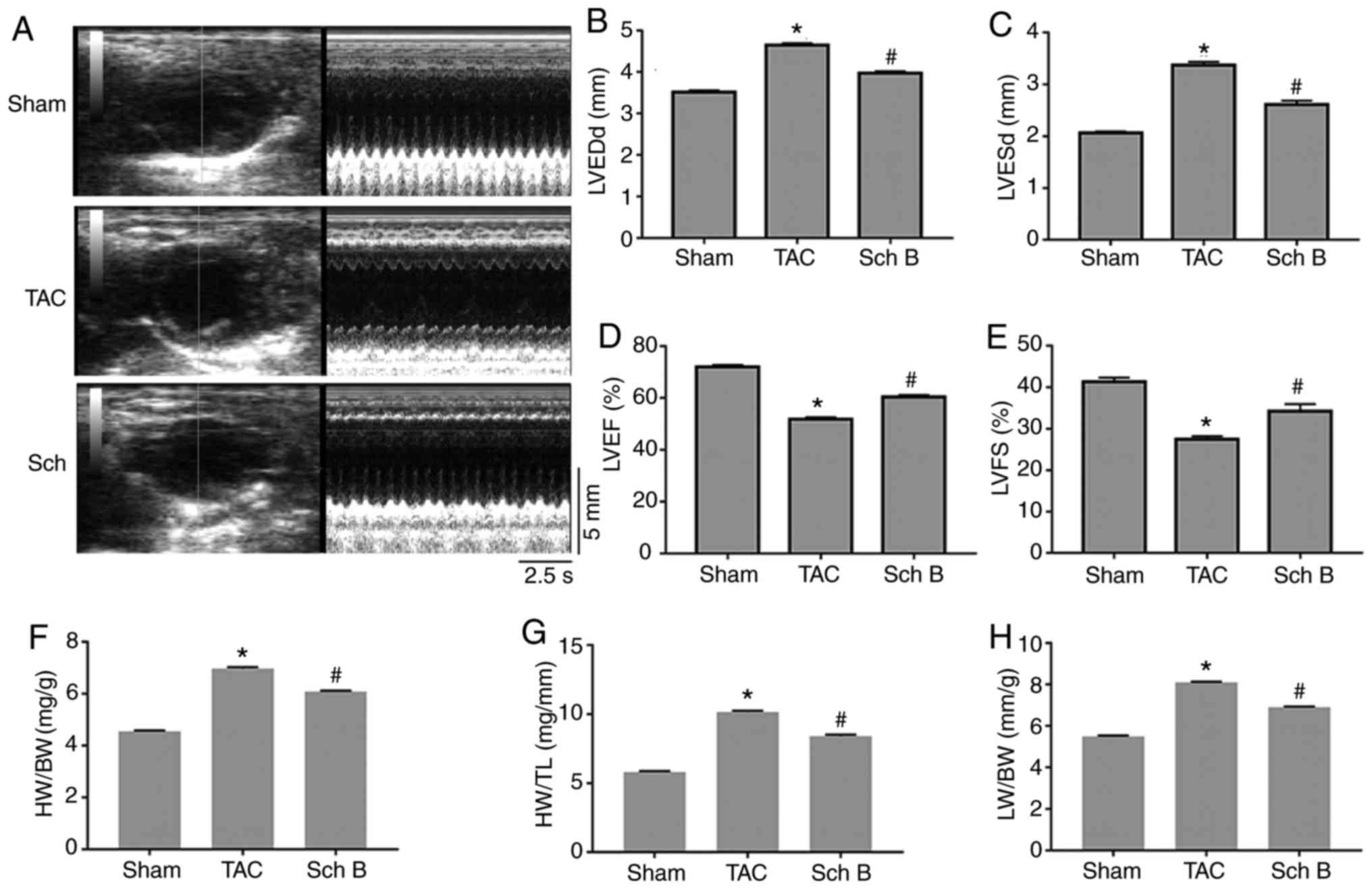 | Figure 1.Cardiac function and structure of the
mice in each experimental group. (A) Representative mouse M-mode
echocardiograms. (B) LVEDd, (C) LVESd, (D) LVEF and (E) FS values
of the mice in each group are presented (n=8). (F) HW/BW, (G) HW/TL
and (H) LW/BW values of the mice in each group are presented (n=6).
Data are expressed as the mean ± SEM. *P<0.05 vs. sham;
#P<0.05 vs. TAC. LVESd, left ventricular end-systolic
diameter; LVEDd, left ventricular end-diastolic diameter; LVEF,
left ventricular ejection fraction; LVFS, left ventricular
fractional shortening; HW, heart weight; BW, body weight; TL, tibia
length; LW, lung weight; TAC, transverse aortic constriction; Sch
B, schisandrin B. |
Cardiac hypertrophy
H&E staining revealed that the cardiomyocyte CSA
in the TAC group was increased compared with that in the sham
group, while that in the Sch B-treated group was significantly
decreased compared with the TAC group (P<0.05; Fig. 2A and B). Similarly, western blotting
demonstrated that the expression levels of cardiac hypertrophy
markers [atrial natriuretic peptide (ANP), brain natriuretic
peptide (BNP) and β-myosin heavy chain (β-MHC)] in the TAC group
were significantly increased compared with the sham group
(P<0.05). The levels of the aforementioned markers were
significantly decreased in the Sch B group compared with the TAC
group (P<0.05; Fig. 2C and
D).
 | Figure 2.Cardiac hypertrophy levels in each
experimental group. (A) Representative H&E staining of left
ventricular tissues obtained from mice in each group
(magnification, ×400). (B) Statistical analysis of the
cardiomyocyte CSA from H&E-stained sections (n=100+
cardiomyocytes in four samples). (C) Representative western
blotting of the cardiac hypertrophy markers ANP, BNP and β-MHC in
each group. (D) Representative statistical analysis of the cardiac
hypertrophy markers ANP, BNP and β-MHC in each group (n=4). Data
are expressed as the mean ± SEM. *P<0.05 vs. respective sham
group; #P<0.05 vs. respective TAC group. H&E,
hematoxylin-eosin; CSA, cross-sectional area; ANP, atrial
natriuretic peptide; BNP, brain natriuretic peptide; β-MHC,
β-myosin heavy chain; TAC, transverse aortic constriction; Sch B,
schisandrin B. |
Myocardial fibrosis
Masson staining revealed that the area of LV
fibrosis in the Sch B-treated group was significantly decreased
compared with that in the TAC group. The staining was significantly
lighter in the Sch B-treated group compared with the TAC group
(P<0.05; Fig. 3A and B).
Similarly, western blotting revealed that the expression levels of
the myocardial fibrosis markers connective tissue growth factor
(CTGF) and collagen I and III in the TAC group were significantly
increased compared with the sham group (P<0.05). The levels of
CTGF and collagen I and III were significantly decreased in the Sch
B group compared with the TAC group (P<0.05; Fig. 3C and D).
Sch B improves LV structural
remodeling by inhibiting the MAPK signaling pathway
The current study investigated the effect of Sch B
on the MAPK signaling pathway during pathological myocardial
remodeling following TAC. The phosphorylation levels of the MAPK
signaling pathway-associated proteins ERK1/2, JNK1/2 and P38 in the
myocardial tissue of the Sch-B-treated animals were significantly
decreased compared with the TAC group (P<0.05; Fig. 4). These data suggested that Sch B
inhibited the MAPK signaling pathway following TAC.
In order to investigate the modulating effect of Sch
B on MAPK signaling, different concentrations of Sch B were used to
pretreat H9c2 cells prior to Ang II stimulation. Cell viability was
determined using MTT assay is presented in Fig. 5A. There was no difference in cell
viability among the different concentrations (0, 5, 10 and 20 µM)
of Sch B. The protein phosphorylation levels of ERK1/2, JNK1/2 and
P38 were increased following Ang II stimulation, which was
significantly suppressed by 10 and 20 µM Sch B compared with the
untreated controls (P<0.05; Fig. 5B
and C). The mRNA levels of the hypertrophy markers ANP, BNP and
β-MHC and the fibrosis mediators CTGF and collagen I and III,
induced by Ang II stimulation, were significantly decreased by 10
and 20 µM Sch B compared with the untreated controls (P<0.05;
Fig. 5D and E). This suggested that
the inactivation of the MAPK signaling pathway reduced the
pathological remodeling induced by Ang II stimulation.
 | Figure 5.Sch B rescues the adverse effects of
Ang II-induced myocyte hypertrophy and fibrosis by inhibiting the
mitogen-activated protein kinase signaling pathway. (A) Cell
viability was determined by MTT assay. (B) Representative western
blotting of ERK1/2, JNK1/2 and P38 in H9c2 cells incubated with
different concentrations of Sch B following exposure to Ang II. (C)
Representative statistical analysis of ERK1/2, JNK1/2 and P38 in
H9c2 cells incubated with different concentrations of Sch B
following exposure to Ang II (n=4). DMSO was used as a control
solution. (D) RT-qPCR analysis of the mRNA levels of ANP, BNP and
β-MHC in H9c2 cells incubated with different concentrations of Sch
B following exposure to Ang II (n=4). DMSO was used as a control
solution. (E) RT-qPCR analysis of the mRNA levels of CTGF and
collagen I and III in H9c2 cells incubated with different
concentrations of Sch B following exposure to Ang II (n=4). DMSO
was used as a control solution. Data are expressed as the mean ±
standard error of the mean. *P<0.05. Sch B, schisandrin B; Ang
II, angiotensin II; ERK, extracellular signal-regulated kinase;
JNK, c-Jun N-terminal kinase; P38, P38 mitogen-activated protein
kinase; RT-qPCR, reverse-transcription quantitative PCR; ANP,
atrial natriuretic peptide; BNP, brain natriuretic peptide; β-MHC,
beta-myosin heavy chain; CTGF, connective tissue growth factor; NS,
not significant; p, phosphorylated. |
Discussion
The results obtained in the current study revealed
that Sch B significantly improved cardiac dysfunction in pressure
overload-induced cardiac remodeling, potentially by decreasing
cardiac hypertrophy and fibrosis. This effect may be due to
inhibition of the MAPK signaling pathway by decreasing the
phosphorylation levels of ERK1/2, JNK1/2 and P38.
Previous studies demonstrated that Sch B has a
protective effect on myocardial injury. Chiu et al (15) demonstrated that Sch B improves
myocardial ischemia-reperfusion injury in rats by inducing the
expression of heat shock protein (HSP) 25 and HSP 70. Moreover, a
previous study also indicated that Sch B ameliorates cardiac
dysfunction in a mouse model of myocardial infarction (14). These observations are consistent with
studies by Thandavarayan et al (13) showing that Sch B prevents
doxorubicin-induced cardiac dysfunction. The present study provides
correlative evidence suggesting that compared with the TAC group,
the LVEF and LVFS in the Sch B-treated group were significantly
increased. Therefore, Sch B may alleviate pressure overload-induced
cardiac dysfunction.
The main pathological changes in pressure overload-
induced pathological myocardial remodeling include myocardial cell
hypertrophy and myocardial fibrosis (23). Cardiomyocyte hypertrophy is one of
the hallmark changes in pathological myocardial remodeling
(24). The present study revealed
that cardiomyocyte CSA was significantly increased in the TAC group
compared with the sham group. Following 4 weeks of treatment with
Sch B, the CSA of cardiomyocytes was significantly reduced compared
with the other groups. Similarly, the Sch B-treated group exhibited
significantly lower levels of cardiac hypertrophy markers compared
with the TAC group. In addition, the HW/BW, HW/TL and LW/BW of the
Sch B-treated group were decreased compared with the TAC group.
Therefore, the results obtained in the current study revealed that
Sch B may improve pressure overload-induced cardiac
hypertrophy.
Myocardial fibrosis is another hallmark of
pathological myocardial remodeling (25). Excessive deposition of cardiac
collagen results in diastolic and contractile dysfunction, leading
to heart failure (26). Previous
studies demonstrated that Sch B attenuates pathological liver,
pulmonary and epidural fibrosis (27–29). A
previous study indicated that Sch B improves cardiac fibrosis in a
mouse model of myocardial infarction (14). Similar results were observed in the
present study. Compared with the TAC group, the Sch B-treated group
exhibited a significantly reduced myocardial fibrosis area, and the
expression of myocardial fibrosis markers (CTGF, collagen I and
9+65 III) was significantly reduced. Therefore, Sch B may
significantly improve myocardial fibrosis caused by TAC.
Previous studies have revealed that the MAPK
signaling pathway is closely associated with cardiac pathology,
including cardiac hypertrophy and myocardial fibrosis (30,31).
Activation of ERK1/2, P38 and JNK1/2 increases cell growth and
migration and promotes myocardial fibrosis (30,31). In
addition, previous studies revealed that Sch B inhibits the MAPK
signaling pathway (13,32,33).
Activation of ERK1/2, JNK1/2 and P38 increases cell growth and
migration and promotes myocardial fibrosis (30). A previous study revealed that Sch B
prevents doxorubicin-induced cardiac damage by inhibiting the MAPK
signaling pathway (10). These
findings are similar to the results of the present study. Compared
with the TAC group, the phosphorylation levels of ERK1/2, JNK1/2
and P38 in the Sch B-treated group were significantly decreased.
This was associated with less cardiac hypertrophy and myocardial
fibrosis in the Sch B-treated group. Moreover, the in vitro
experiments in the current study further suggested that Sch B
alleviated Ang II-induced cardiac fibrosis and hypertrophy by
decreasing the phosphorylation levels of ERK1/2, JNK1/2 and P38.
Similarly, a number of previously published studies demonstrated
that Sch B prevents cardiac damage via suppression of the MAPK
signal pathway (13,31,32).
The current study had a number of limitations. The
present study revealed that Sch B may prevent pressure
overload-induced cardiac fibrosis and hypertrophy by inhibition of
the MAPK signaling pathway. Future experiments should use small
interfering RNA or inhibitors of the MAPK pathway to further verify
the role of MAPK in the protective effect of Sch B. Whether the
MAPK signaling pathway is a direct target of Sch B remains unclear.
Further studies are required to elucidate the mechanism by which
Sch B regulates the MAPK signaling pathway.
In conclusion, the present study demonstrated that
Sch B may improve pathological myocardial remodeling and cardiac
function induced by pressure overload through inhibition of the
MAPK signaling pathway.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FA, QHG, XG and ZC designed the study. FA, QHG and
BY performed the experiments. FA, QHG and WL performed data
analysis and interpreted the data. FA, QHG, BY and WL built the
model and analyzed data. XG and ZC wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The use of animals in the present study was reviewed
and approved by the Ethics Committee of the Central Hospital of
Wuhan (Wuhan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Metra M and Teerlink JR: Heart failure.
Lancet. 390:1981–1995. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burchfield JS, Xie M and Hill JA:
Pathological ventricular remodeling: Mechanisms: Part 1 of 2.
Circulation. 128:388–400. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Opie LH, Commerford PJ, Gersh BJ and
Pfeffer MA: Controversies in ventricular remodelling. Lancet.
367:356–367. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Drazner MH: The progression of
hypertensive heart disease. Circulation. 123:327–334. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Houser SR, Margulies KB, Murphy AM,
Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin
JD, Sussman MA, et al: Animal models of heart failure: A scientific
statement from the american heart association. Circ Res.
111:131–150. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shimizu I and Minamino T: Physiological
and pathological cardiac hypertrophy. J Mol Cell Cardiol.
97:245–262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nature
reviews. Nat Rev Mol Cell Biol. 7:589–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tham YK, Bernardo BC, Ooi JY, Weeks KL and
McMullen JR: Pathophysiology of cardiac hypertrophy and heart
failure: Signaling pathways and novel therapeutic targets. Arch
Toxicol. 89:1401–1438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Panossian A and Wikman G: Pharmacology of
schisandra chinensis bail: An overview of Russian research and uses
in medicine. J Ethnopharmacol. 118:183–212. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ip SP, Poon MK, Wu SS, Che CT, Ng KH, Kong
YC and Ko KM: Effect of schisandrin B on hepatic glutathione
antioxidant system in mice: Protection against carbon tetrachloride
toxicity. Planta Med. 61:398–401. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Checker R, Patwardhan RS, Sharma D, Menon
J, Thoh M, Bhilwade HN, Konishi T and Sandur SK: Schisandrin B
exhibits anti-inflammatory activity through modulation of the
redox-sensitive transcription factors Nrf2 and NF-κB. Free Radic
Biol Med. 53:1421–1430. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu Y, Liu Z, Sun J, Pan Q, Sun F, Yan Z
and Hu X: Schisandrin B prevents doxorubicin-induced chronic
cardiotoxicity and enhances its anticancer activity in vivo. PLoS
One. 6:e283352011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thandavarayan RA, Giridharan VV, Arumugam
S, Suzuki K, Ko KM, Krishnamurthy P, Watanabe K and Konishi T:
Schisandrin B prevents doxorubicin induced cardiac dysfunction by
modulation of DNA damage, oxidative stress and inflammation through
inhibition of MAPK/p53 signaling. PLoS One. 10:e01192142015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen P, Pang S, Yang N, Meng H, Liu J,
Zhou N, Zhang M, Xu Z, Gao W, Chen B, et al: Beneficial effects of
schisandrin B on the cardiac function in mice model of myocardial
infarction. PLoS One. 8:e794182013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chiu PY and Ko KM: Schisandrin B protects
myocardial ischemia-reperfusion injury partly by inducing Hsp25 and
Hsp70 expression in rats. Mol Cell Biochem. 266:139–144. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang W, Sun Z and Meng F: Schisandrin B
ameliorates myocardial ischemia/reperfusion injury through
attenuation of endoplasmic reticulum stress-induced apoptosis.
Inflammation. 40:1903–1911. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Y, Jiang XL, Liu Y, Jiang DS, Zhang Y,
Zhang R, Chen Y, Yang Q, Zhang XD, Fan GC and Li H:
Toll-interacting protein (Tollip) negatively regulates pressure
overload-induced ventricular hypertrophy in mice. Cardiovasc Res.
101:87–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Z, You L, Cheng Y, Hu K, Wang Z,
Cheng Y, Yang J, Yang Y and Wang G: Investigation of
pharmacokinetics, tissue distribution and excretion of schisandrin
B in rats by HPLC-MS/MS. Biomed Chromatogr. 32:2018.
|
|
19
|
Ko KM, Chen N, Leung HY, Leong EP, Poon MK
and Chiu PY: Long-term schisandrin B treatment mitigates
age-related impairments in mitochondrial antioxidant status and
functional ability in various tissues, and improves the survival of
aging C57BL/6J mice. Biofactors. 34:331–342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leong PK, Chiu PY and Ko KM:
Prooxidant-induced glutathione antioxidant response in vitro and in
vivo: A comparative study between schisandrin B and curcumin. Biol
Pharm Bull. 35:464–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chiu PY and Ko KM: Schisandrin B-induced
increase in cellular glutathione level and protection against
oxidant injury are mediated by the enhancement of glutathione
synthesis and regeneration in AML12 and H9c2 cells. Biofactors.
26:221–230. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Beetz N, Rommel C, Schnick T, Neumann E,
Lother A, Monroy-Ordonez EB, Zeeb M, Preissl S, Gilsbach R,
Melchior-Becker A, et al: Ablation of biglycan attenuates cardiac
hypertrophy and fibrosis after left ventricular pressure overload.
J Mol Cell Cardiol. 101:145–155. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhai M, Liu Z, Zhang B, Jing L, Li B, Li
K, Chen X, Zhang M, Yu B, Ren K, et al: Melatonin protects against
the pathological cardiac hypertrophy induced by transverse aortic
constriction through activating PGC-1beta: In vivo and in vitro
studies. J Pineal Res. 63:2017. View Article : Google Scholar
|
|
25
|
Moens AL, Takimoto E, Tocchetti CG, Chakir
K, Bedja D, Cormaci G, Ketner EA, Majmudar M, Gabrielson K,
Halushka MK, et al: Reversal of cardiac hypertrophy and fibrosis
from pressure overload by tetrahydrobiopterin: Efficacy of
recoupling nitric oxide synthase as a therapeutic strategy.
Circulation. 117:2626–2636. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Urban ML, Manenti L and Vaglio A:
Fibrosis-A common pathway to organ injury and failure. N Engl J
Med. 373:95–96. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen Q, Zhang H, Cao Y, Li Y, Sun S, Zhang
J and Zhang G: Schisandrin B attenuates CCl4-induced
liver fibrosis in rats by regulation of Nrf2-ARE and TGF-β/Smad
signaling pathways. Drug Des Devel Ther. 11:2179–2191. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang D, Liu B, Cao B, Wei F, Yu X, Li GF,
Chen H, Wei LQ and Wang PL: Synergistic protection of Schizandrin B
and Glycyrrhizic acid against bleomycin-induced pulmonary fibrosis
by inhibiting TGF-β1/Smad2 pathways and overexpression of NOX4. Int
Immunopharmacol. 48:67–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jin H, Wang Z, Gu Z, Wu J, Bai X, Shao Z,
Miao J, Wang Q, Wang Q and Wang X: Schisandrin B attenuates
epidural fibrosis in postlaminectomy rats by inhibiting
proliferation and extracellular matrix production of fibroblasts.
Phytother Res. 33:107–116. 2019. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hunter JJ, Tanaka N, Rockman HA, Ross J Jr
and Chien KR: Ventricular expression of a MLC-2v-ras fusion gene
induces cardiac hypertrophy and selective diastolic dysfunction in
transgenic mice. J Biol Chem. 270:23173–23178. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cowan KJ and Storey KB: Mitogen-activated
protein kinases: New signaling pathways functioning in cellular
responses to environmental stress. J Exp Biol. 206:1107–1115. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ran J, Ma C, Xu K, Xu L, He Y, Moqbel SAA,
Hu P, Jiang L, Chen W, Bao J, et al: Schisandrin B ameliorated
chondrocytes inflammation and osteoarthritis via suppression of
NF-κB and MAPK signal pathways. Drug Des Devel Ther. 12:1195–1204.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park EJ, Chun JN, Kim SH, Kim CY, Lee HJ,
Kim HK, Park JK, Lee SW, So I and Jeon JH: Schisandrin B suppresses
TGFbeta1 signaling by inhibiting Smad2/3 and MAPK pathways. Biochem
Pharmacol. 83:378–384. 2012. View Article : Google Scholar : PubMed/NCBI
|
















