Introduction
Primary liver cancer is a common malignant tumor,
ranking sixth in the global incidence of cancer and fourth in tumor
mortality in 2018(1). Liver cancer
is characterized by high rates of recurrence and high metastasis,
severely hindering the overall efficacy of the treatment methods
for this disease (2).
Embryonic stem cells (ESCs) are derived from the
inner cell mass of blastocyst and have self-renewal, unlimited
proliferation and potential differentiation abilities (3). Some types of tumor cells can form
teratomas in vivo that are insensitive to growth signal
inhibition and cell death stimuli (4), suggesting that cancer and ESCs share
similar phenotypes in terms of cell differentiation, proliferation
and cell invasion. In addition, normal tadpoles could be obtained
by transplanting the nucleus of a Lucke's kidney cancer cell into a
denuclearized fertilized egg, where no tumor tissue was formed
(3). A study by Illmensee (5) demonstrated that other embryonic tissue
cells, except for embryonic cells, do not possess this ability, and
indicated that tumor cells can also be induced to differentiate
into mature histiocytes under specific conditions.
The tumor microenvironment, consisting of
microvasculature, extracellular matrix and various stromal cells
(tumor-associated fibroblasts, mesenchymal stem cells and
endothelial cells) and signaling molecules secreted by these cells,
play an important role in the process of tumorigenesis, development
and metastasis (5,6). ESC conditioned medium (ESC-CM) could be
used to simulate the ESC microenvironment in vitro (7).
Giuffrida et al (7) revealed that ESC-CM can inhibit the
proliferation of ovarian cancer cells by regulating the cell cycle,
which was associated with the secretion of small molecules by ESCs.
The ability of ESC-CM to inhibit the proliferation and invasion of
tumor cells is associated with the secretion of lefty A by ESCs
(8). The proliferation of breast
cancer is also inhibited in ESC-CM (9). ESC-CM resulted in decreased cancer cell
migration, invasion, angiogenesis and decreased the ability of
tumor formation following subcutaneous transplantation in mice. The
antitumor effects of ESC-CM were mediated by inhibition of tumor
cell proliferation, angiogenesis, migration, and STAT3 signaling
pathway (8).
Exosomes serve important roles in extracellular
signal transduction in both tumor and normal cells (10), which includes a number of bioactive
substances such as heat shock proteins and microRNAs (miRNAs)
(11). miRNAs are endogenous small
RNAs ~20-24 nucleotides in length and have important regulatory
functions in the cell. miRNAs are formed by multi-step digestion in
cells, which involves the formation of pri-miRNA, pre-miRNA and
finally mature miRNA. miRNA 290-295 in the exosomes derived from
ESCs, particularly miRNA 294, have been shown to ameliorate
myocardial infarction in mice (12).
miRNA 294 was demonstrated to improve myocardial angiogenesis and
myocardial cell viability, and decrease myocardial fibrosis,
following myocardial infarction.
The inoculation of animals with ESCs can effectively
prevent the occurrence of colon (9),
lung (10) and ovarian cancer
(11). ESCs have therapeutic effects
on early tumors with low tumor burden and can effectively decrease
the incidence of inflammation-associated tumors (13); however, the underlying mechanisms are
unknown.
To date, the regulation of tumor cell miRNAs by
ESC-CM has been poorly investigated (12). In the present study, ESCs and
hepatocellular carcinoma Hepal-6 cells were co-cultured via
non-direct contact, in order to investigate the inhibitory effect
of ESC-CM on the biological behavior of liver tumor cells in
vitro. By comparing the tumor cell miRNA expression profile
between ESC-CM treatment and mouse embryonic fibroblast (MEF)-CM
treatment, the possible miRNAs underlying the regulatory mechanisms
were explored. The findings of the present study can help determine
the association between miRNAs and the malignant behaviors of
tumors.
Materials and methods
Materials
MTT was obtained from Sigma-Aldrich (Merck KGaA) and
Transwell chambers with 0.4-µm pore sizes were purchased from
Corning Inc. Cell cycle and apoptosis analysis (cat. no. C1052) and
Annexin V-Phycoerythrin Apoptosis Detection Kits (cat. no. C1065L)
were purchased from Beyotime Institute of Biotechnology. Antibodies
against β-actin, cyclin-dependent kinase (CDK)2, CDK4, CDK6, cyclin
D1 and cyclin E1 were purchased from Cell Signaling Technology,
Inc.
Cell lines and culture conditions
ESCs and MEFs were supplied by Cyagen Biosciences,
Inc. MEFs were cultured in the media of mouse embryonic fibroblast
basal medium, 10% FBS, 1% glutamine and 100 U/ml
penincillin-streptomycin. The C57BL/6 ESCs were cultured on plates
pre-coated with gelatin solution, irradiated C57BL/6 MEFs as feeder
cells and mouse ESCs medium (mESC basal medium, 15% fetal bovine
serum, penincillin-streptomycin, 1% glutamine, nonessential amino
acid, 1,000 U/ml leukemia inhibitory factor, 0.1 mM
2-mercaptoethanol; all medium obtained from Cyagen Bioscience
Inc.). Hepa1-6 cells were purchased from the Cell Bank of Type
Culture Collection of the Chinese Academy of Sciences, maintained
in Dulbecco's modified Eagle's medium (DMEM) with high glucose
supplemented with 10% heat-inactivated FBS (both obtained from
Gibco; Thermo Fisher Scientific, Inc.) at 37˚C in a humidified
atmosphere containing 5% CO2.
CM culture
ESC-CM was obtained by overlaying MEF cells with
ESCs in the aforementioned mouse ESC growth medium. for 24, 48 or
72 h (days 1, 2 and 3 ESC-CM). Control CM was made by incubating
MEFs with stem cell medium for 24 48 or 72 h (day 1, 2 and 3
MEF-CM). Feeder and ESCs were cultured in 90-mm plates containing
10 ml stem cell medium. Feeders were plated at 8x105
cells per plate and ESCs were plated at 2x105 cells per
plate. CM was harvested and passed through a syringe filter to
remove any cellular debris.
Hepa1-6 and ESCs co-culture
The 24-well Transwell chambers (pore size, 0.4 µm;
membrane diameter, 6.5 mm) were purchased from Corning, Inc. When
the pore size of the co-culture system was <3.0 µm, the cells
could not pass through. In this co-culture system, ESCs were seeded
into the lower chamber whilst the Hepal-6 cells were seeded into
the upper chamber. Hepa1-6 cells were plated in 2 ml DMEM medium
supplemented with 10% heat-inactivated FBS at a density of
5x105 cells/well in the chamber. ESCs on feeder cells
and feeder cells only were separately plated at a density of
1x105 cells in culture plate, where was under chamber.
These cells were allowed to attach overnight. Following 24 h
incubation, DMEM was replaced by stem cell medium and chambers with
feeders only or ESCs on feeders were placed in wells containing
cancer cells. Co-culture occurred for 24-144 h wherein medium was
changed every 24 h. The cell number was assessed at 24, 48, 72, 96,
120 and 144 h using the MTT method.
Cell proliferation assay
The effect of ESC-CM on Hepa1-6 cell proliferation
was measured by MTT assay. Cells were plated in 96-well plates at a
density of 2,500 cells/well overnight, following which they were
treated with day 1, 2 and 3 ESC-CM and MEF-CM. Following incubation
for 24, 48, 72, 96, 120 or 144 h at 37˚C in a humidified incubator,
MTT (5 mg/ml in PBS) was added to each well and incubated for 4 h.
Subsequently, the medium was removed and 0.1 ml of buffered DMSO
was added per well. The absorbance was recorded on a microplate
reader (Sepctra Max M2e; Molecular Devices, LLC) at the wavelength
of 490 nm. The proliferation curve was generated with time on the
horizontal axis and OD value on the vertical axis.
Cell cycle analysis
Following treatment with CM, the DNA content and
cell cycle distribution of Hepa1-6 cells were determined by flow
cytometry. Cells plated at a density of 5x105 cells/well
in six-well plates were treated with ESC-CM or MEF-CM for 48 h and
harvested at 24, 48, 72 or 96 h. The cells were washed in PBS and
then fixed in cold 70% ethanol and stored at 4˚C for 30 min. The
cells were washed with cold PBS and resuspended in PBS solution
containing 50 µg/ml phycoerythrin (PE) and 100 U/ml of RNase type
A. Cells were then incubated in the dark for 30 min at 37˚C. Cell
cycle was analyzed by flow cytometry (BD Biosciences) and the
FlowJo 7.6.1 software (version 7.6.1; FlowJo LLC).
Quantification of apoptosis
For apoptosis analysis, 5x105 Hepa1-6
cells/well were plated on six-well plates and treated with ESC-CM
or MEF-CM for 48 h and harvested at 24, 48, 72 or 96 h. The cells
were labeled with annexin V and PE. Apoptosis rates were determined
by flow cytometry (BD Biosciences) and analyzed using the FlowJo
software. The percentage of early apoptosis was calculated by
counting annexin V-positive and PI-negative cells, and the
percentage of the late apoptosis was calculated using annexin
V-positive and PI-positive cells.
Western blotting
Hepa1-6 cells treated with MEF-CM or ESC-CM for 72 h
were lysed in RIPA buffer (150 mM NaCl, 0.5% sodium deoxycholate,
0.1% SDS, 1% NP40, 1 mM EDTA and 50 mM Tris pH 8.0). The protein
content was determined using the Bicinchoninic Acid protein assay
kit (Beyotime Institute of Biotechnology). Equivalent amounts of
protein (50 µg) were separated by 10% SDS-PAGE gel and transferred
to polyvinylidenedifluoride (PVDF) membranes. The membranes were
incubated with blocking buffer (5% nonfat dry milk) for 2 h at 4˚C
and then incubated with primary antibodies against Cyclin D1
(1:1,000; cat. no. 26939-1-AP; Proteintech Group, Inc.), Cyclin E1
(1:1,000; cat. no. 11554-1-AP; Proteintech Group, Inc.), CDK2
(1:1,000; cat. no. 2546T; Cell Signaling Technology, Inc.), CDK4
(1:1,000; cat. no. 12790T; Cell Signaling Technology, Inc.), CDK6
(1:1,000; cat. no. 13331T; Cell Signaling Technology, Inc.) or
β-actin (1:5,000; cat. no. A5441; Sigma Aldrich; Merck KGaA)
overnight at 4˚C. The membranes were then incubated with
horseradish peroxidase-conjugated anti-mouse (1:5,000; cat. no.
HAF007, R&D systems, Inc.) or anti-rabbit (1:5,000; cat. no.
HAF008; R&D systems, Inc.) IgG for 1 h at 37˚C. The blots were
subsequently detected using chemiluminescence (BeyoECL plus kit,
Beyotime Institute of Biotechnology) and analyzed by Image J
version 18.0 (National Institutes of Health).
Reverse transcription-quantitative-PCR
(RT-qPCR)
Total RNA was obtained from cancer cells cultured
with ESC-CM for 48 h or MEF-CM for 72 h using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) or miRNeasy
mini kit (Qiagen GmbH) according to corresponding manufacturers'
protocols. Quantification was performed using a two-step reaction
process: Reverse transcription (RT) and PCR. Each RT reaction
consisted of 1 µg RNA, 4 µl miScript HiSpec Buffer, 2 µl nucleic
acid Mix and 2 µl miScript Reverse Transcriptase Mix (Qiagen GmbH),
in a total volume of 20 µl per reaction. Reactions were performed
in a GeneAmp® PCR System 9700 (Applied Biosystems;
Thermo Fisher Scientific, Inc.) for 60 min at 37˚C, followed by
heat inactivation of RT for 5 min at 95˚C. The 20 µl RT reaction
mix was then diluted 5X in nuclease-free water and held at -20˚C.
Subsequent qPCR was performed using LightCycler® 480 II
Real-time PCR Instrument (Roche Diagnostics) with 10 µl PCR
reaction mixture that included 1 µl cDNA, 5 µl 2X
LightCycler® 480 SYBR Green I Master (Roche
Diagnostics), 0.2 µl universal primers (Qiagen GmbH), 0.2 µl
microRNA-specific primers and 3.6 µl nuclease-free water. The
thermocycling conditions were as follows: Initial denaturation at
95˚C for 10 min, followed by 40 cycles of 95˚C for 10 sec and 60˚C
for 30 sec. Each sample was run in triplicate. At the end of the
PCR cycles, melting curve analysis was performed to validate the
specific generation of the expected PCR product. The
microRNA-specific primer sequences were designed in the laboratory
and synthesized by Generay Biotech Co., Ltd. based on the microRNA
sequences obtained from the miRBase database (ftp://mirbase.org/pub/mirbase/20/), which were
listed in Table I. The expression
levels of microRNAs were normalized to U6) and were calculated
using the 2-ΔΔCq method (14).
 | Table IPrimer sequences used for reverse
transcription-quantitative PCR. |
Table I
Primer sequences used for reverse
transcription-quantitative PCR.
| Primer name | Primer sequence
(5'-3') |
|---|
| U6 |
CAAGGATGACACGCAAATTCG |
| Mmu-miR-10a-5p |
TACCCTGTAGATCGAATTTGTG |
| Mmu-miR-1187 |
TATGTGTGTGTGTATGTGTGTAA |
| Mmu-miR-134-5p |
TGTGACTGGTTGACCAGAGGGG |
|
Mmu-miR-29b-1-5p |
GCTGGTTTCATATGGTGGTTTA |
|
Mmu-miR-3070b-3p |
TGGTGCTATGGTCAGGGGTAGA |
| Mmu-miR-421-3p |
ATCAACAGACATTAATTGGGCGC |
miRNA experiment and data
analysis
Total RNA was extracted with TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.) and miRNeasy mini kit
(Qiagen GmbH). RNA levels were quantified by the
NanoDrop™ 2000 (Thermo Fisher Scientific, Inc.) and the
RNA integrity was assessed using Agilent Bioanalyzer 2100 (Agilent
Technologies, Inc.). The sample labeling, microarray hybridization
and washing were performed based on the manufacturer's standard
protocols. Briefly, total RNA was dephosphorylated, denatured and
then labeled with Cyanine-3-CTP using Low Input QuickAmp Labeling
Kit, one-Color (cat. no. 5190-2305; Agilent technologies, Inc.).
Following purification, the labeled RNAs were hybridized onto
SurePrint Mouse microRNA microarrays (cat. no. G4872A-046065;
Agilent technologies, Inc.). After washing, the arrays were scanned
with the Agilent Scanner G2505C (Agilent Technologies, Inc.). The
associated differentially expressed miRNA primer sequences that
were used for qPCR are provided in Table
I.
The Feature Extraction software (version 10.7.1.1;
Agilent Technologies, Inc.) was used to analyze array images to
obtain raw data. The Genespring software (version 12.5; Agilent
Technologies, Inc.) was used to complete the basic analysis with
the raw data. Initially, the raw data were normalized with the
quantile algorithm. The probes with at least 100% of samples in any
one condition out of two conditions and flagged as ‘detected’ were
chosen for further data analysis. Differentially expressed miRNAs
were subsequently identified through fold change as well as P-value
calculated using t-test. The threshold set for upregulated and
downregulated genes was a fold change >2.0 and a P<0.05. The
target genes of differentially expressed miRNAs were the
intersection of miRNAs predicted by three databases (Targetscan
version 6.0; http://www.targetscan.org; microRNAorg version 6.2,
http://www.microrna.org; Pita release 2010,
https://omictools.com/pita-tool). Gene
Ontology (GO, release number 2019-07-01; http://geneontology.org/) and Kyoto Encyclopedia of
Genes and Genomes (KEGG, release number 2019-10-01; https://www.kegg.jp/) analysis were applied to
determine the roles of these target genes. Hierarchical clustering
was performed to show the distinguishable miRNA expression pattern
among samples.
Statistical analysis
Data are presented as the mean ± standard deviation.
SPSS 16.0 (SPSS, Inc.) and GraphPad Prism 6.0 (GraphPad Software,
Inc.) were used for the statistical analysis and graphical display
of data, respectively. Each experiment was repeated three times.
The differences between groups were analyzed by Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ESCs secrete factors that inhibit
Hepa1-6 proliferation
To determine whether mouse ESCs secreted factors
that inhibit Hepa1-6 proliferation, Transwell chambers were used to
perform co-culture experiments. The chambers separated ESCs from
Hepa1-6 cells by a 0.4-µm pore, high-density membrane, which
allowed factors secreted by ESC to diffuse to Hepa1-6 cells,
without any direct contact.
Hepa1-6 cells (1x106) were co-cultured
with ESC for 144 h and MTT assays were performed every 24 h. By 72
h, compared with MEFs (control group), ESCs caused inhibition of
Hepa1-6 cell proliferation, and the effect was also observed at 96,
120 and 144 h (Fig. 1A). To
determine the effect of ESC microenvironment on Hepa1-6 cell
proliferation, day 1 2 and 3 ESC-CM treatment and MEF-CM treatment
were used for the MMT assay. The media in which Hepa1-6 cells were
seeded into 96-well plates and incubated for 24 h was replaced by
day 1 2 or 3 ESC-CM or MEF-CM. MTT assays were performed every 24
h. Day 1 ESC-CM suppressed Hepa1-6 cell proliferation only at 120
and 144 h; however day 2 ESC-CM inhibited cell proliferation at 72,
96, 120 and 144 h (Fig. 1B and
D). Furthermore, the day 3 ESC-CM
resulted in stronger inhibition than day 2 ESC-CM, with a
significant inhibition of cell proliferation at 48, 72, 96, 120 and
144 h (Fig. 1C).
ESC-CM arrests cells at G1
phase of the cell cycle
In order to understand how ESC-CM inhibits Hepa1-6
cell proliferation, cell cycle analysis was performed. Cells were
treated with day 2 ESC-CM or MEF-CM (control group) and harvested
following 24-96 h and stained with PI for fluorescence-activated
cell sorting analysis. The results indicated an increased Hepa1-6
cell number in G1 phase from 54.99±0.95 (24 h ESC-CM) to
68.83±0.18% (96 h ESC-CM) (P<0.001) and a reduction in cell
numbers in G2/M phase from 20.03±0.38% (24 h ESC-CM) to
7.26±0.16% (96 h ESC-CM) (P<0.001; Fig. 2). These findings indicate that ESC-CM
inhibited cell proliferation by arresting cells at G1
phase of the cell cycle.
Effect of ESC-CM on cell
apoptosis
In order to further investigate whether ESC-CM could
inhibit cell proliferation through increased apoptosis, Hepa1-6
cells were treated with day 2 ESC-CM or MEF-CM for 24-96 h prior to
harvesting. The cells were stained with Annexin V-FITC and PI and
were analyzed by flow cytometry. There were no significant
differences in the number of early apoptotic cells (1.0±0.09%) at
24 h and in the number of late apoptotic cells (1.2±0.03%) at 96 h
in cells treated with ESC-CM compared with MEF-CM (early apoptotic
cells, 0.6±0.07%; late apoptotic cells, 1.1±0.03%) at the end of
the experiment. These data indicate that the ESC-CM did not mediate
its growth inhibitory effects through increased apoptosis (Fig. 3).
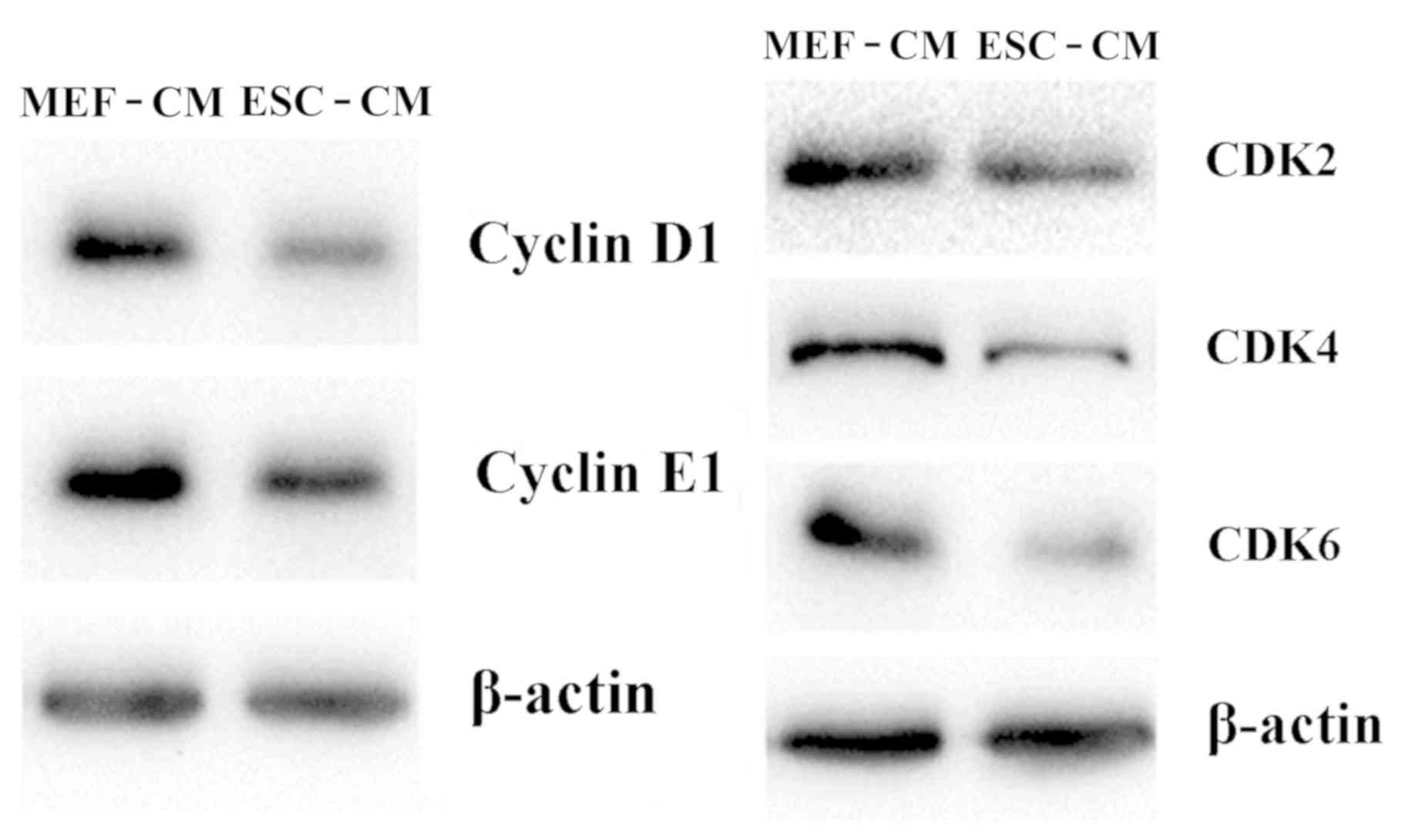
ESC-CM downregulates the expression of
G1 phase-associated CDKs
The mechanism of arresting cells at the
G1 phase by ESC-CM was investigated by determining the
expression of CDKs, including Cyclin D1, Cyclin E1, CDK2, CDK4 and
CDK6. As shown in Fig. 4, treatment
with day 2 ESC-CM for 72 h resulted in downregulated cellular
protein expression of Cyclin D1, CDK2, Cyclin E1, CDK4 and CDK6
compared with Hepa1-6 cells in MEF-CM for 72 h. These findings
suggest that ESC-CM arrested Hepa1-6 cells in the G1
phase of the cell cycle by downregulating the expression of
G1 phase-associated cyclin D1 and CDKs.
miRNA profile
In order to further explore the effect of ESC-CM on
miRNA expression in Hepa1-6 cells, the miRNA profile of Hepal-6
cells cultured with 72-h ESC-CM were compared with the control
group co-cultured with 72-h MEF-CM. A total of 6 differentially
expressed miRNAs were found in the ESC-CM group, specifically three
miRNAs (miR-29b-1-5p, miR-10a-5p and miR-421-3p) were upregulated
and three miRNAs (miR-1187, miR-134-5p and miR-3070b-3p) were
downregulated (Fig. 5A). These
results were subsequently confirmed by RT-qPCR analysis (Fig. 5B).
A total of 423 predicted target genes were obtained
according to the differentially expressed miRNAs (Fig. 6). These target genes were
particularly enriched in the ‘cell cycle’ (CCND2, CDK4, CDK14 and
Notch2), ‘apoptotic process’ and ‘signal transduction’ (Fig. 7). The results of the KEGG analysis
revealed the ‘Wnt signaling pathway’ and the ‘Hippo signaling
pathway’ to be particularly enriched (Fig. 8).
Discussion
To date, the mechanism of tumor occurrence,
development, metastasis and recurrence is not completely
elucidated, which is a complex biological process and associated
with genomic instability, chromosomal abnormalities and genetic
mutations. With the in-depth study of tumors, it was found that the
occurrence, development and metastasis were associated with the
surrounding environment of tumors (13). There is a biphasic and dynamic
interaction between the tumor microenvironment and tumor cells
(15). Studies have shown that the
embryonic microenvironment can alter the biological behavior of
malignant tumor cells, and that cytokines secreted by ESCs regulate
the proliferation and invasion of some tumor cells (15,16). In
addition, owing to fewer ESC antigens being expressed in normal
tissues compared with traditional cancer vaccines, ESCs cause fewer
autoimmune responses while killing tumor cells (17).
In the present study, 24-h CM from ESCs was
demonstrated to inhibit the proliferation of hepatocellular
carcinoma cells after 120 h, whereas 72-h CM inhibited the
proliferation of hepatocellular carcinoma cells at 48 h. All CM
inhibited the proliferation of HCC cells, but the inhibitory
ability of CM at different co-culture durations was different,
which may be associated with the concentration of bioactive
substances secreted by ESCs. Furthermore, ESC-CM blocked
hepatocellular carcinoma cells at the G1 phase, whereas
no significant differences were observed in the apoptosis of
Hepa1-6 cells. Furthermore, the expression levels of
G1-associated regulatory proteins (cyclin D1/CDK4/CDK6)
in the ESC-CM group were significantly lower compared with that in
the MEF-CM group.
In order to explore the mechanism further, six
significantly differentially expressed miRNAs were identified in
the group with 48-h ESC-CM treatment compared with the group with
72-h MEF-CM treatment. Furthermore, 423 putative target genes of
the regulated miRNAs were predicted. Among them, miR-134-5p has
been reported to function as a tumor suppressor gene in gastric
cancer (18), and inhibit
proliferation and promote apoptosis of lung cancer cells by
inhibiting the ERK1/2 signaling pathway (19). High expression of miR-29b-1-5p was
demonstrated to inhibit tumor cell proliferation and migration in
breast cancer (20). miR-1187 may
play a role in viral hepatitis (21). In a previous study, miR-421-3p was
abnormally expressed in post-traumatic stress disorder (22). miR-10a-5p can function as an oncogene
or a tumor suppressor gene depending on the cell type, which
demonstrated that miR-10a-5p had tumor-specific and time-space
specificities (16-18).
To the best of our knowledge the biological function of
miR-3070b-3p was not reported in the previous literature.
The KEGG analysis revealed the importance of the Wnt
signaling pathway in the occurrence and development of liver
cancer. In a previous study, Wnt/β-catenin signaling pathway was an
important factor in the early development and progression of liver
cancer (23). Furthermore, some
studies have shown that non-canonical Wnt signaling pathway can
inhibit the proliferation and metastasis of hepatoma cells by
antagonizing the classical Wnt signaling pathway (24,25).
CDK14 was highly expressed in hepatocarcinoma tissues, which was
associated with the invasion function of hepatoma cells (26). CDK14 can regulate cell cycle and cell
proliferation by interacting with cyclin D3 and cyclin Y (27,28). The
complexes of CDK14 and cyclin Y can activate the non-canonical Wnt
signaling pathway in liver cancer cells (29). In addition, the Hippo signaling
pathway also plays an important role in cell proliferation,
apoptosis, cell cycle and differentiation (30). The screening for effective miRNAs
that regulate the Wnt signaling pathway may be a new approach for
liver cancer treatment.
In conclusion, ESC-CM was demonstrated to regulate
the expression of cyclin D1/CDK4/CDK6 and the Wnt and Hippo
signaling pathways through miRNAs, and resulted in cell cycle
arrest at the G1 phase and inhibited cell proliferation
of Hepa1-6 cells.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the author on reasonable request.
Authors' contributions
LL and YZ designed the study and carried out the
experiments. LL, YZ and QZ performed the analysis. JJ participated
in the design of the study. All authors read and approved the final
version of the manuscript.
Ethical approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ryan MJ, Willatt J, Majdalany BS, Kielar
AZ, Chong S, Ruma JA and Pandya A: Ablation techniques for primary
and metastatic liver tumors. World J Hepatol. 8:191–199.
2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Boroviak T, Loos R, Bertone P, Smith A and
Nichols J: The ability of inner-cell-mass cells to self-renew as
embryonic stem cells is acquired following epiblast specification.
Nat Cell Biol. 16:516–528. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Williams JW III, Carlson DL, Gadson RG,
Rollins-Smith L, Williams CS and Mckinnell RG: Cytogenetic analysis
of triploid renal carcinoma in Rana pipiens. Cytogenet Cell Genet.
64:18–22. 1993.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Illmensee K: Reversion of malignancy and
normalized differentiation of teratocarcinoma cells in chimeric
mice. Basic Life Sci. 12:3–25. 1978.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Melzer C, von der Ohe J, Lehnert H,
Ungefroren H and Hass R: Cancer stem cell niche models and
contribution by mesenchymal stroma/stem cells. Mol Cancer.
16(28)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Giuffrida D, Rogers IM, Nagy A, Calogero
AE, Brown TJ and Casper RF: Human embryonic stem cells secrete
soluble factors that inhibit cancer cell growth. Cell Prolif.
42:788–798. 2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cavallari C, Fonsato V, Herrera MB, Bruno
S, Tetta C and Camussi G: Role of lefty in the anti tumor activity
of human adult liver stem cells. Oncogene. 32:819–826.
2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
He N, Feng G, Li Y, Xu Y, Xie X, Wang H,
Wang Y, Ou L, Pei X and Liu N: Embryonic stem cell preconditioned
microenvironment suppresses tumorigenic properties in breast
cancer. Stem Cell Res Ther. 7(95)2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Becker A, Thakur BK, Weiss JM, Kim HS,
Peinado H and Lyden D: Extracellular vesicles in cancer:
Cell-to-cell mediators of metastasis. Cancer Cell. 30:836–848.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Gopal SK, Greening DW, Rai A, Chen M, Xu
R, Shafiq A, Mathias RA, Zhu HJ and Simpson RJ: Extracellular
vesicles: Their role in cancer biology and epithelial-mesenchymal
transition. Biochem J. 474:21–45. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Khan M, Nickoloff E, Abramova T, Johnson
J, Verma SK, Krishnamurthy P, Mackie AR, Vaughan E, Garikipati VN,
Benedict C, et al: Embryonic stem cell-derived exosomes promote
endogenous repair mechanisms and enhance cardiac function following
myocardial infarction. Circ Res. 117:52–64. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yaddanapudi K, Mitchell RA, Putty K,
Willer S, Sharma RK, Yan J, Bodduluri H and Eaton JW: Vaccination
with embryonic stem cells protects against lung cancer: Is a
broad-spectrum prophylactic vaccine against cancer possible? PLoS
One. 7(e42289)2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang ZJ, Chen XH, Chang XH, Ye X, Li Y
and Cui H: Human embryonic stem cells-a potential vaccine for
ovarian cancer. Asian Pac J Cancer Prev. 13:4295–4300.
2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang Z, Chen X, Chang X, Ye X, Li Y and
Cui H: Vaccination with embryonic stem cells generates effective
antitumor immunity against ovarian cancer. Int J Mol Med.
31:147–153. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Adam V, Wauters I and Vansteenkiste J:
Melanoma-associated antigen-A3 vaccination in the treatment of
non-small-cell lung cancer. Exp Opin Biol Ther. 14:365–376.
2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Qiu ZA and He GP: MicroRNA-134 functions
as a tumor suppressor gene in gastric cancer. Am J Transl Res.
8:4320–4328. 2016.PubMed/NCBI
|
|
19
|
Chen T, Gao F, Feng S, Yang T and Chen M:
MicroRNA-134 regulates lung cancer cell H69 growth and apoptosis by
targeting WWOX gene and suppressing the ERK1/2 signaling pathway.
Biochem Biophys Res Commun. 464:748–754. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Muluhngwi P, Krishna A, Vittitow SL,
Napier JT, Richardson KM, Ellis M, Mott JL and Klinge CM: Tamoxifen
differentially regulates miR-29b-1 and miR-29a expression depending
on endocrine-sensitivity in breast cancer cells. Cancer Lett.
388:230–238. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yu DS, An FM, Gong BD, Xiang XG, Lin LY,
Wang H and Xie Q: The regulatory role of microRNA-1187 in
TNF-α-mediated hepatocyte apoptosis in acute liver failure. Int J
Mol Med. 29:663–668. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Balakathiresan NS, Chandran R, Bhomia M,
Jia M, Li H and Maheshwari RK: Serum and amygdala microRNA
signatures of posttraumatic stress: Fear correlation and biomarker
potential. J Psychiatr Res. 57:65–73. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liu LJ, Xie SX, Chen YT, Xue JL, Zhang CJ
and Zhu F: Aberrant regulation of Wnt signaling in hepatocellular
carcinoma. World J Gastroenterol. 22:7486–7499. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yuzugullu H, Benhaj K, Ozturk N, Senturk
S, Celik E, Toylu A, Tasdemir N, Yilmaz M, Erdal E, Akcali KC, et
al: Canonical Wnt signaling is antagonized by noncanonical Wnt5a in
hepatocellular carcinoma cells. Mol Cancer. 8(90)2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Toyama T, Lee HC, Koga H, Wands JR and Kim
M: Noncanonical Wnt11 inhibits hepatocellular carcinoma cell
proliferation and migration. Mol Cancer Res. 8:254–265.
2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Pang EY, Bai AH, To KF, Sy SM, Wong NL,
Lai PB, Squire JA and Wong N: Identification of PFTAIRE protein
kinase 1, a novel cell division cycle-2 related gene, in the motile
phenotype of hepatocellular carcinoma cells. Hepatology.
46:436–445. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Shu F, Lv S, Qin Y, Ma X, Wang X, Peng X,
Luo Y, Xu BE, Sun X and Wu J: Functional characterization of human
PFTK1 as a cyclin-dependent kinase. Proc Natl Acad Sci USA.
104:9248–9253. 2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jiang M, Gao Y, Yang T, Zhu X and Chen J:
Cyclin Y, a novel membrane-associated cyclin, interacts with PFTK1.
FEBS Lett. 583:2171–2178. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sun T, Co NN and Wong N: PFTK1 interacts
with cyclin Y to activate non-canonical Wnt signaling in
hepatocellular carcinoma. Biochem Biophys Res Commun. 449:163–168.
2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang H, Du YC, Zhou XJ, Liu H and Tang SC:
The dual functions of YAP-1 to promote and inhibit cell growth in
human malignancy. Cancer Metastasis Rev. 33:173–181.
2014.PubMed/NCBI View Article : Google Scholar
|