Introduction
Colorectal cancer (CRC) is the third most commonly
diagnosed cancer type worldwide. The incidence and death rates of
CRC continue to decline in individuals aged ≥50 years but continues
to rise in those aged <50 years (1). In contrast to the survival rate of ~65%
for patients with localized disease, the 5-year survival rate of
patients with metastatic CRC after diagnosis is <10% (2). Hence, disease detection and treatment
at an early stage are crucial (3).
Considering the relatively high prevalence in the population, novel
treatment strategies must be developed by characterizing the
genetic, epigenetic, transcriptomic and proteomic changes to
improve disease detection and surveillance. Epigenetic alterations
have been established as a hallmark of tumorigenesis (4,5). DNA
methylation, as an important epigenetic process, is a common
molecular alteration in CRC (6) and
contributes to the later stages of colorectal carcinogenesis
(7). Thus, DNA methylation of
multiple promoters may serve as a biomarker for the early detection
and monitoring of CRC progression. However, DNA methylation of
genes and its consequences in CRC remain poorly characterized. In
addition to global hypomethylation, genome-scale studies have
yielded important insight into promoter methylation, including the
CpG-island methylator phenotype (CIMP), which displays extensive
DNA hypermethylation at a unique set of CpG islands (8,9). The
Cancer Genome Atlas (TCGA) database (http://tcga-data.nci.nih.gov/tcga/) is a comprehensive
molecular database of cancer genomes and epigenomes and has
provided an excellent opportunity for analyses on molecular
diagnostics for cancer (10,11).
In the present study, methylation-regulated genes
were investigated by integrating epigenetic and genomic data from
the TCGA database. Pearson's correlation between gene methylation
and expression was determined and the genes with R>0.4 were
regarded as methylated genes. In addition, methylation-regulated
genes associated with the prognosis of patients with CRC (key
genes) were identified by combining the data from the TCGA
database. Furthermore, the key gene was validated in the Cancer
Cell Line Encyclopedia (CCLE) database (https://portals.broadinstitute.org/ccle) (12). The CpG island methylation of key
genes was further evaluated. The results demonstrated that zinc
finger (ZNF)726, which was regulated by DNA methylation at the CpG
islands in its promoter region, may be an independent prognostic
risk factor for overall survival (OS) of patients with CRC.
Materials and methods
Screening of methylated genes in the
TCGA database
The transcriptome profiling and DNA methylation data
(IlIumina Human Methylation 450) were downloaded from the TCGA
database. Finally, the data of 428 tissues (407 colorectal tumor
tissues and 21 adjacent non-tumor tissues) were obtained, which
contained RNA expression data and DNA methylation information. For
gene methylation vs. expression, Pearson's correlation coefficients
of >0.4 and P<0.05 were considered to indicate
methylation-regulated genes, and these were further studied.
Patients without clinical information, including age, gender, TNM
stage, residual tumor reaction margin and survival, were excluded.
Finally, 390 patients with CRC were enrolled in the clinical
relevance study.
Pathway analysis
Pathway enrichment analysis of DNA-methylated genes
screened from TCGA was performed on the basis of the consensus
PathDB database (http://cpdb.molgen.mpg.de/) (13). Over-representation analysis (14) was utilized for the DNA-methylated
gene list. The following pathway databases were defined for the
analysis: Manual upload, NetPath, Signalink, PID, EHMN, HumanCyc,
INOH, KEGG, Biocarta, WikiPathways, SMPDB and PharmGKB (13). Minimum overlap input list >2 and
P<0.05 indicated statistical significance.
DNA-methylated genes predicting the
survival of patients with CRC
To evaluate whether the expression of certain
methylated genes was able to predict patient survival, 390 patients
with CRC were divided into high and low expression groups based on
the median of gene expression. The OS and disease-free survival
(DFS) of patients in two subgroups for each gene were evaluated
using Kaplan-Meier survival curves and the log-rank test.
Prognosis-associated genes (key genes) were then obtained. It was
inferred that if any of the key genes was an independent predictive
factor for OS of CRC patients, the clinicopathologic features were
analyzed using univariate and multivariable Cox proportional
hazards models. The hazard ratio and 95% confidence interval (CI)
were thereby determined. The diagnostic value of DNA-methylated
genes in patients with CRC was further evaluated by using receiver
operating characteristic (ROC) curve analysis. The area under the
ROC curve (AUC) was subsequently calculated.
Experimental validation in the CCLE
database
To compare the role of the ZNF726 gene in CRC with
that in other cancer types, the expression and methylation levels
of key genes were examined in 13 other types of tumor tissues from
the TCGA database. Furthermore, 21 colorectal cancer cell lines
from the CCLE database (https://portals.broadinstitute.org/ccle/about) were
selected to detect the expression and methylation levels and their
relevance was verified. The expression and methylation levels of
key genes in tissues and cell lines were determined.
CpG island methylation of a key gene
and its clinical value
To continue detecting the methylation levels of the
promoter of the key gene, the CpG islands were downloaded and
analyzed. The Pearson correlation between key gene expression and
CpG island methylation was also evaluated. Finally, the methylation
profile of CpG islands was further verified in Gene Expression
Omnibus (GEO) datasets (https://www.ncbi.nlm.nih.gov/gds/).
Statistical analysis
The methylation and expression data from the TCGA
were analyzed with R 3.5.1 software (https://www.r-project.org/) using MethylMix version
3.7(15) and limma version 3.363
packages. The Kaplan-Meier survival analysis was performed using
the survival package in R. Differences between the groups were
assessed via Student's t-test and visualized by ggstatsplot
(16) version 0.05 and ggplot2
version 3.0.0(17) in R. The
univariate/multivariate Cox proportional hazard regression analysis
was performed using SPSS version 22 (IBM, Corp.). The association
of global/CpG site methylation with the gene expression level was
estimated by Pearson's correlation analysis. P<0.05 was
considered to indicate statistical significance.
Results
Screening of methylated genes in the
TCGA database
To study the methylation regulation in CRC,
transcriptome profiling and DNA methylation data were downloaded
from the TCGA. A total of 428 samples (21 normal and 407 tumor
tissues) containing expression and methylation data were included
in the study. Subsequently, the Pearson correlation between the
methylation and expression levels was assessed and genes with a
correlation coefficient of >0.4 were regarded as methylated
genes. A total of 177 methylation-regulated genes were thereby
obtained. The expression and methylation levels and Pearson
correlations were calculated and presented in Fig. 1 and Table
SI.
Pathway analysis
Pathway enrichment analysis was performed to explore
the possible mechanism of methylation-regulated genes in CRC. Gene
expression, genomic transcription pathway and RNA polymerase II
transcription were prominent pathways. The top 20 pathways are
provided in Fig. 2.
Methylation-regulated ZNF726 predicts
the OS and DFS of patients with CRC
On the basis of the median value of differentially
expressed genes, 390 patients with CRC were divided into high and
low expression groups and the Kaplan-Meier survival curves were
drawn. As a result, three key genes (NXPE4, REP15 and ZNF726),
which were able to effectively distinguish the patients' prognosis,
were screened. Patients with high expression of neurexophilin and
PC-esterase domain family member 4 (NXPE4), RAB15 effector protein
(REP15) and ZNF726 had a significantly improved prognosis regarding
OS (Fig. 3A). However, only the
expression of ZNF726 was able to predict the DFS of patients with
CRC (Fig. 3B). The three genes were
significantly less expressed and hypermethylated in tumor tissues
(Fig. 3C and D). The significant correlation between
methylation and expression levels presented in Fig. 4A and B
further suggested their important role in tumors.
 | Figure 4DNA methylation of three key genes,
NXPE4, REP15 and ZNF726. (A) Pearson correlation between the
expression and methylation of three prognosis-associated genes. (B)
Summary of methylation of the genes. The histograms display the
distribution of methylation among the tumor samples and the
horizontal black bars represent the methylation values in the
normal samples. The mixture components 1 and 2 represent the peak
of methylation. (C) ROC curves of the three genes methylation for
early diagnosis of patients with CRC from The Cancer Genome Atlas
database. Exp, expression; methy, methylation; ROC, receiver
operating characteristic; AUC, area under the ROC curve; R, Pearson
correlation coefficient; REP15, RAB15 effector protein; ZNF, zinc
finger protein; NXPE4, neurexophilin and PC-esterase domain family
member 4. |
Subsequently, ROC curves were plotted to evaluate
the diagnostic value of NXPE4, REP15 and ZNF726 methylation
(Fig. 4C) between tumor and normal
tissues. The AUCs for the three genes were 0.773, 0.792 and 0.929,
respectively. The sensitivity and specificity were (1.000, 0.518),
(1.000, 0.634) and (1.000, 0.860), respectively.
Cox regression analysis was performed to further
verify whether ZNF726 may be used as an independent prognostic risk
factor for OS in patients with CRC. Univariate Cox analysis
suggested that age (P<0.001), TNM stage (P<0.001), T stage
(P=0.003), N stage (P<0.001), M stage (P<0.001), residual
tumor (P<0.001) and ZNF726 expression (P=0.024) were associated
with prognosis and multivariate Cox regression analysis suggested
that age (P=0.005), TNM stage (P<0.001), T stage (P=0.014), N
stage (P=0.036), M stage (P=0.035), residual tumor (P=0.042),
ZNF726 expression (P=0.015) were independent prognostic risk
factors for OS in patients with CRC (Fig. 5).
Experimental validation in cancer
tissues and CRC cell lines
The expression and methylation of ZNF726 were
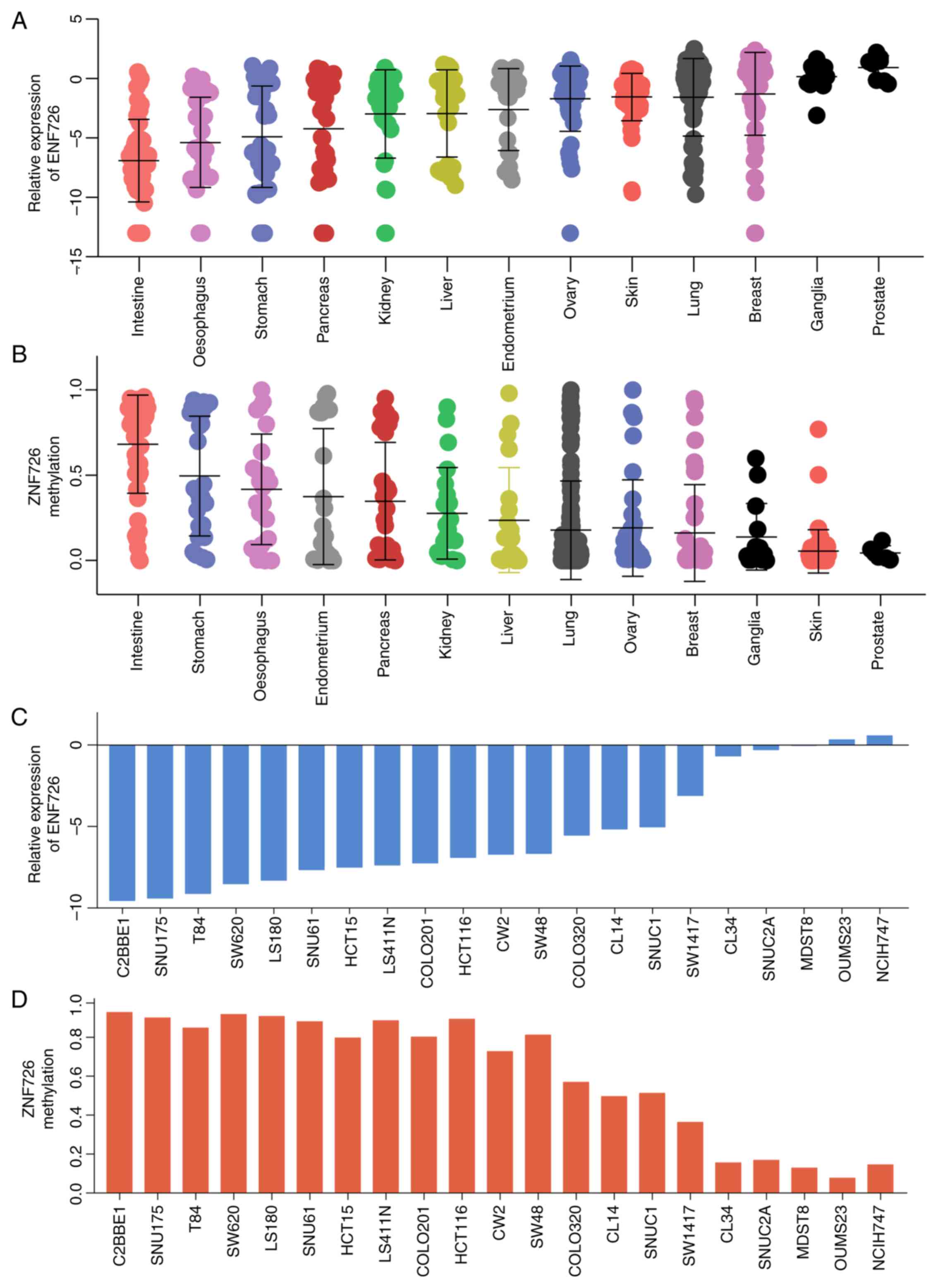
further examined in various types of tumor tissue. As presented in
Fig. 6A and B, among 13 tumor tissue types, ZNF726 had
the lowest expression and highest methylation in CRC, indicating
that the role of ZNF726 in CRC may be linked to methylation
regulation. Similarly, the expression and methylation levels of
ZNF726 were negatively correlated in 21 CRC cell lines (Fig. 6C and D). These results further confirmed that
ZNF726 was downregulated in CRC and regulated by DNA
methylation.
CpG island methylation analysis of
ZNF726
To further determine the mode of regulation of gene
expression via methylation of the gene promoter, the CpG island
methylation dataset was downloaded from the TCGA database. Only one
CpG island (cg20649823) in the ZNF726 promoter was obtained. As
presented in Fig. 7A, the
methylation level of cg20649823 was significantly different between
tumor and normal tissues. Furthermore, cg20649823 methylation and
ZNF726 expression exhibited a significant correlation (R=-0.527;
Fig. 7B). The methylation level of
cg20649823 was also assessed in datasets from the GEO database
(GSE42752, GSE75546 and GSE77965). As presented in Fig. 7C, based on the methylation level of
cg20649823, it was possible to clearly distinguish CRC from normal
tissues.
Discussion
CRC is characterized by the accumulation of genetic
and epigenetic alterations in colonic epithelial cells in
neoplastic processes. Cancer-specific genetic and epigenetic
aberrations may be detected for non-invasive cancer diagnosis and
monitoring (18). Aberrant
hypermethylation of DNA at CpG islands, which mediates the
transcriptional silencing of tumor suppressor genes, is an early
event integral to gastrointestinal cancer development (3). Studies on the genetic and epigenetic
modifications of molecular markers in human cancers in the TCGA
database have opened new avenues for cancer detection
approaches.
In the present study, CRC genomic and epigenetic
data from the TCGA database were integrated to characterize the DNA
methylation landscape in CRC. It was inferred that the methylated
genes targeted by epigenetic alterations in tumors may have an
important role in tumor initiation and progression. Pathway
enrichment analysis revealed prominent pathways, including those of
transcription and metabolism, in tumorigenesis. Numerous known
cancer-associated genes were included in the methylated gene set of
the present study, certain of which are known to cause tumor
development. For instance, the ectopic expression of transforming
growth factor beta induced is associated with enhanced metastasis
and extravasation of CRC (19-21). Polymeric
immunoglobulin receptor promotes tumor growth in hepatocellular
carcinoma (22), but inhibits
pancreatic and periampullary adenocarcinoma development (23). Homeobox (HOX) expression contributes
to CRC development: HOXB3, HOXB8 and HOXB9 are significantly
upregulated, whereas HOXB2 and HOXB13 are significantly
downregulated in carcinoma tissues (24). Furthermore, the aberrant regulation
of bone marrow stromal cell antigen 2 is associated with breast
cancer (25), hepatic carcinoma
(26), esophageal cancer, gastric
cancer and CRC (27,28). The epigenetic regulation of E74-like
ETS transcription factor 5 is associated with the metastasis of
breast cancer (29) and urothelial
cancer (30).
Considering the clinical significance of methylation
regulatory genes, Kaplan-Meier survival analysis was performed and
the candidate key genes (NXPE4, REP15 and ZNF726), whose aberrant
expression was linked to CRC prognosis, screened. ZNF proteins,
including ZNF346, ZNF638, ZNF700 and ZNF768, are capture antigens
for detecting autoantibodies in CRC (31). In addition, DNA methylation
biomarkers of the ZNF gene family, combined with other genes, may
be useful in the clinical setting for head and neck squamous cell
carcinoma (32). Consistent with
previous studies, the current study identified 44 ZNF genes
(Table SI) that are prominently
regulated by methylation, further indicating the significance of
the ZNF family in CRC development. Detection in multiple cancer
types indicated that ZNF726 was the least expressed and most
methylated in CRC, implying the high relevance of its methylation
and marked oncogenic potential. Two novel genes (NXPE4 and REP15)
associated with CRC prognosis require further research.
Analysis of the consequences of CIMPs on gene
expression suggested that only a small percentage (7%) of gene
expression was downregulated with high CIMP in tumors. However,
this downregulation appears to be enriched for genes essential for
the genesis of CRC (33). The
epigenetic driver ZNF726 identified in the present study featured
CIMP DNA hypermethylation and reduced gene expression. The
epigenetic driver genes are attractive therapeutic targets,
considering that their activation by treatment with DNA methylation
inhibitors may result in CRC occurrence. However, the present study
lacks further clinical validation. In a follow-up study, the
consistency of the level of ZNF726 in peripheral blood and CRC
tissue will be further evaluated.
In conclusion, the present study presented a
detailed knowledge base on the genes with altered DNA methylation
and its associated pathways in CRC, paving the way for the
development of novel cancer therapies. Epigenetically activated
ZNF726 was identified as a novel prognosis-associated gene and may
become a novel target for CRC treatment.
Supplementary Material
Table SI. Methylated genes and their
methylation profile compared between colorectal cancer tumor and
non-tumor tissue.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Jiangsu
Provincial Key Research Development Program (grant no. BE2016794 to
JF and grant no. BE2016795 to JW).
Availability of data and materials
All data and materials are available from the TCGA
(http://tcga-data.nci.nih.gov/tcga/)
and GEO databases https://www.ncbi.nlm.nih.gov/gds/).
Authors' contributions
HZ contributed to statistical analysis and
manuscript preparation. XS and YL contributed to manuscript editing
and the interpretation of data. JW and JF contributed to the design
of the current study. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ,
Meester RGS, Barzi A and Jemal A: Colorectal cancer statistics,
2017. CA Cancer J Clin. 67:177–193. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Weisenberger DJ, Liang G and Lenz HJ: DNA
methylation aberrancies delineate clinically distinct subsets of
colorectal cancer and provide novel targets for epigenetic
therapies. Oncogene. 37:566–577. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wong CC, Li W, Chan B and Yu J: Epigenomic
biomarkers for prognostication and diagnosis of gastrointestinal
cancers. Semin Cancer Biol. 55:90–105. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Du Z, Fei T, Verhaak RG, Su Z, Zhang Y,
Brown M, Chen Y and Liu XS: Integrative genomic analyses reveal
clinically relevant long non-coding RNAs in human cancer. Nat
Struct Mol Biol. 20:908–913. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tse JWT, Jenkins LJ, Chionh F and
Mariadason JM: Aberrant DNA methylation in colorectal cancer: What
should we target? Trends Cancer. 3:698–712. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kim MS, Lee J and Sidransky D: DNA
methylation markers in colorectal cancer. Cancer Metastasis Rev.
29:181–206. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wang Z, Yang B, Zhang M, Guo W, Wu Z, Wang
Y, Jia L, Li S, Cancer Genome Atlas Research Network , Xie W
and Yang D: lncRNA epigenetic landscape analysis identifies EPIC1 as
an oncogenic lncRNA that interacts with MYC and promotes cell-cycle
progression in cancer. Cancer Cell. 33:706–720.e9. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Toyota M, Ahuja N, Ohe-Toyota M, Herman
JG, Baylin SB and Issa JP: CpG island methylator phenotype in
colorectal cancer. Proc Natl Acad Sci USA. 96:8681–8686.
1999.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer. Nature
487: 330.337, 2012.
|
|
11
|
Cancer Genome Atlas Research Network:
Comprehensive molecular characterization of gastric adenocarcinoma.
Nature 513: 202.209, 2014.
|
|
12
|
Barretina J, Caponigro G, Stransky N,
Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV,
Sonkin D, et al: The cancer cell line encyclopedia enables
predictive modelling of anticancer drug sensitivity. Nature.
483:603–607. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kamburov A, Wierling C, Lehrach H and
Herwig R: ConsensusPathDB-a database for integrating human
functional interaction networks. Nucleic Acids Res. 37:D623–D628.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sanford T, Meng MV, Railkar R, Agarwal PK
and Porten SP: Integrative analysis of the epigenetic basis of
muscle-invasive urothelial carcinoma. Clin Epigenetics.
10(19)2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gevaert O: MethylMix: An R package for
identifying DNA methylation-driven genes. Bioinformatics.
31:1839–1841. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Indrajeet Patil: ‘ggplot2’ Based Plots
with Statistical Details (R package ggstatsplot version 0.0.1).
2018.
|
|
17
|
Wickham H: Ggplot2: Elegant Graphics for
Data Analysis. J R Stat Soc. 174:245–246. 2011.
|
|
18
|
Xu RH, Wei W, Krawczyk M, Wang W, Luo H,
Flagg K, Yi S, Shi W, Quan Q, Li K, et al: Circulating tumour DNA
methylation markers for diagnosis and prognosis of hepatocellular
carcinoma. Nat Mater. 16:1155–1161. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Ma C, Rong Y, Radiloff DR, Datto MB,
Centeno B, Bao S, Cheng AW, Lin F, Jiang S, Yeatman TJ and Wang XF:
Extracellular matrix protein betaig-h3/TGFBI promotes metastasis of
colon cancer by enhancing cell extravasation. Genes Dev.
22:308–321. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yokobori T and Nishiyama M: TGF-β
signaling in gastrointestinal cancers: Progress in basic and
clinical research. J Clin Med. 6(E11)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang H, Dong S and Feng J: Epigenetic
profiling and mRNA expression reveal candidate genes as biomarkers
for colorectal cancer. J Cell Biochem. 120:10767–10776.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yue X, Ai J, Xu Y, Chen Y, Huang M, Yang
X, Hu B, Zhang H, He C, Yang X, et al: Polymeric immunoglobulin
receptor promotes tumor growth in hepatocellular carcinoma.
Hepatology. 65:1948–1962. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Richard Fristedt, Jacob Elebro, Alexander
Gaber, Björn Nodin, Mathias Uhlén and Karin Jirström: Abstract B30:
High expression of PIGR is an independent favorable prognostic
factor in pancreatic and periampullary adenocarcinoma. Cancer Res.
75:B30. 2015.
|
|
24
|
Bhatlekar S, Fields JZ and Boman BM: HOX
genes and their role in the development of human cancers. J Mol Med
(Berl). 92:811–823. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Cai D, Cao J, Li Z, Zheng X, Yao Y, Li W
and Yuan Z: Up-regulation of bone marrow stromal protein 2 (BST2)
in breast cancer with bone metastasis. BMC Cancer.
9(102)2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Pan XB, Qu XW, Jiang D, Zhao XL, Han JC
and Wei L: BST2/Tetherin inhibits hepatitis C virus production in
human hepatoma cells. Antiviral Res. 98:54–60. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chiang SF, Kan CY, Hsiao YC, Tang R, Hsieh
LL, Chiang JM, Tsai WS, Yeh CY, Hsieh PS, Liang Y, et al: Bone
marrow stromal antigen 2 is a novel plasma biomarker and
prognosticator for colorectal carcinoma: A secretome-based
verification study. Dis Markers. 2015(874054)2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Mukai S, Oue N, Oshima T, Mukai R,
Tatsumoto Y, Sakamoto N, Sentani K, Tanabe K, Egi H, Hinoi T, et
al: Overexpression of transmembrane protein BST2 is associated with
poor survival of patients with esophageal, gastric, or colorectal
cancer. Ann Surg Oncol. 24:594–602. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Gallego-Ortega D, Ledger A, Roden DL, Law
AM, Magenau A, Kikhtyak Z, Cho C, Allerdice SL, Lee HJ, Valdes-Mora
F, et al: ELF5 drives lung metastasis in luminal breast cancer
through recruitment of Gr1+ CD11b+
myeloid-derived suppressor cells. PLoS Biol.
13(e1002330)2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yao B, Zhao J, Li Y, Li H, Hu Z, Pan P,
Zhang Y, Du E, Liu R and Xu Y: Elf5 inhibits TGF-β-driven
epithelial-mesenchymal transition in prostate cancer by repressing
SMAD3 activation. Prostate. 75:872–882. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
O'Reilly JA, Fitzgerald J, Fitzgerald S,
Kenny D, Kay EW, O'Kennedy R and Kijanka GS: Diagnostic potential
of zinc finger protein-specific autoantibodies and associated
linear B-cell epitopes in colorectal cancer. PLoS One.
10(e0123469)2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Gaykalova DA, Vatapalli R, Wei Y, Tsai HL,
Wang H, Zhang C, Hennessey PT, Guo T, Tan M, Li R, et al: Outlier
analysis defines zinc finger gene family DNA methylation in tumors
and saliva of head and neck cancer patients. PLoS One.
10(e0142148)2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hinoue T, Weisenberger DJ, Lange CP, Shen
H, Byun HM, Van Den Berg D, Malik S, Pan F, Noushmehr H, van Dijk
CM, et al: Genome-scale analysis of aberrant DNA methylation in
colorectal cancer. Genome Res. 22:271–282. 2012.PubMed/NCBI View Article : Google Scholar
|