Introduction
Ischemia/reperfusion (I/R) and hypoxia/reoxygenation
(H/R) injuries are harmful to endothelial cells in the arterial
system, and are the primary causes of cardiac artery disease (CAD)
(1). Reperfusion and reoxygenation
induce reactive oxygen species (ROS) accumulation and mitochondrial
dysfunction, leading to apoptotic cell death (2). Therefore, therapeutic drugs and
molecules that decrease or prevent reperfusion/reoxygenation injury
have been investigated, which has improved the knowledge of the
pathophysiology of the processes and has exhibited encouraging
results in preclinical trials (3,4).
Glycyrrhizic acid (GA), a triterpene saponin
glycoside, is the primary bioactive component of the root extract
of Glycyrhiza glabra (licorice) (5). Although it is clinically used as an
antiulcer, antiallergic, antioxidant, antiviral and anticancer
agent (6), several studies have also
demonstrated its potential protective effects against I/R- and
H/R-induced endothelial injury (6,7). Cai
et al (8), reported that GA
elicits protective effects against myocardial I/R injury by
regulating oxidative stress and inflammatory reactions via the
transcriptional modification of high-mobility group box 1 and
mitogen-activated protein kinase in rats (8). However, whether GA exhibits protective
effects against I/R- and H/R-induced injury in coronary artery
endothelial cells (CAECs) is not completely understood.
Mitochondria are essential eukaryotic organelles
that are the primary source of cellular energy and participate in
essential physiological processes, including cell signaling and
apoptosis (5-7).
During I/R or H/R, ROS accumulation decreases the mitochondrial
permeability transition, decreases the mitochondrial membrane
potential and alters mitochondrial homeostasis, which particularly
affects myocardial and endothelial cells (9). The accumulation of excessive ROS
critically damages mitochondria, resulting in damage to DNA, lipids
and proteins (10). Damaged
mitochondria subsequently undergo mitophagy, which results in
decreased ATP production, impaired calcium buffering and
ultimately, apoptosis (4,11). ROS accumulation also promotes
autophagy/mitophagy to remove the damaged mitochondria, leading to
further mitochondrial dysfunction (12), which has been reported to be closely
associated with H/R-induced CAD.
To evaluate the effects of GA, a model of H/R injury
was constructed with human CAECs (HCAECs) using a
hypoxia/reoxygenation system. The present study aimed to
investigate whether GA affected ROS accumulation and subsequent
mitochondrial dysfunction; therefore, indicating whether GA may
display protective effects against H/R-induced CAD.
Materials and methods
Cell culture and establishment of the
H/R model
HCAECs were purchased from iCell Bioscience, Inc.
(www.icellbioscience.com/cellDetail/914/0/-1) and
cultured in Endothelial Cell Medium (Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS (Thermo Fisher Scientific, Inc.) at
37˚C with 5% CO2.
To mimic ischemia, hypoxia should be induced in
oxygen-free and nutrition-free conditions; therefore, HCAECs were
cultured with pure nitrogen for 30 min at 37˚C to expel the air and
subsequently, pure nitrogen gas was used to fill the culture
vessels and hypoxia chamber (Corning Inc.). Subsequently, HCAECs
were cultured in hypoxic solution in the hypoxia chamber for 4 h at
37˚C. Endothelial cell medium (cat. no. CC-3162; Lonza Group, Ltd.)
containing 10% FBS (cat. no. 10099; Thermo Fisher Scientific, Inc.)
was pre-maintained in a hypoxia chamber at 37˚C for 24 h. Following
hypoxia induction, the medium was replaced with oxygenated culture
medium supplemented with 10% FBS and the culture vessels were
transferred into a normoxic incubator at 37˚C with 5%
CO2 for 2 h of reoxygenation. To evaluate the effect of
GA on H/R, 50, 100, 150 or 200 µM GA was added to culturing medium
immediately after H/R exposure and incubated for 1 h at 37˚C. After
4, 8 or 12 h, cell viability was measured by performing CCK-8 assay
as described below.
To scavenge total ROS, 10 µM NAC was added into
endothelial cell medium containing 10% FBS together with H/R
exposure. To scavenge mitochondrial ROS, 1 µM MitoQ10
(Sigma-Aldrich; Merck KGaA) was added into medium together with H/R
exposure.
To inhibit autophagy/mitophagy, 100 µmol/l rapamycin
was added into endothelial cell medium containing 10% FBS together
with H/R exposure. To inhibit LC3B-II degradation, 20 µmol/l
chloroquine (Sigma-Aldrich; Merck KGaA) was added into endothelial
cell medium containing 10% FBS together with H/R exposure.
Evaluation of cell viability
Cells (5x103) were seeded into a 96-well
plate and incubated overnight 37˚C. To evaluate cell viability
after hypoxia treatment for 4, 8 or 12 h, 10 µl Cell Counting Kit-8
(CCK-8) solution (Shanghai Shenggong Biology Engineering Technology
Serve, Ltd.) was added to each well and incubated at 37˚C for 2 h.
The absorbance of each well was measured at a wavelength of 450 nm
using a Multiskan spectrum microplate reader (Thermo Fisher
Scientific, Inc.).
Apoptosis assay
Apoptotic cells were detected using the Annexin V/PI
Apoptosis kit (BioVision, Inc.) following the manufacturer's
protocol. Briefly, cell suspensions were made using 0.25% trypsin
and the cell concentration was modified to 1x106
cells/ml. Subsequently, cells (5x105) were stained with
5 µl Annexin V-FITC and 10 µl propidium iodide (PI) for 10 min in
darkness at room temperature. Apoptotic cells were detected using a
3 laser Navios flow cytometer (Beckman Coulter, Inc.). The Annexin
V-FITC positive/PI negative (early stage apoptosis) and Annexin
V-FITC positive/PI positive (late stage apoptosis) subpopulations
were considered as apoptotic cells. Data were analyzed using FlowJo
software (version 10.5.2; FlowJo LLC).
Detecting intracellular and
mitochondrial ROS levels in HCAECs
To measure intracellular and mitochondrial ROS
levels, 2',7'-dichlorofluorescin diacetate (DCFH-DA) and MitoSOX™
staining were performed, respectively. Nuclei were counterstained
with DAPI to a final concentration of 5 µg/ml (Beyotime Institute
of Biotechnology) at room temperature for 5 min. Cells were washed
3 times with ice-cold PBS. Subsequently, cells were incubated with
1 ml serum-free medium containing 10 µM DCFH-DA probe or 5 µM
MitoSOX™ in the dark at 37˚C for 30 min with gentle shaking every 5
min. Cells were washed 3 times with ice-cold PBS. Green
fluorescence was observed using an X71 (U-RFL-T) fluorescence
microscope (Olympus Corporation) at an excitation wavelength of 488
nm and magnification, x40.
JC-1 staining
JC-1 staining (Thermo Fisher Scientific, Inc.) was
used to investigate the mitochondrial membrane potential. Cells
were stained with 15 µg/ml JC-1 at 37˚C for 30 min in dark.
Subsequently, cells were washed 3 times with PBS. Stained cells
were observed using an X71 (U-RFL-T) fluorescence microscope
(Olympus Corporation) at a magnification, x100.
ATP measurement
To assess ATP production, the ATP Bioluminescent
assay kit (Sigma-Aldrich; Merck KGaA) was used according to the
manufacturer's protocol. Cells were washed 3 times with ice-cold
PBS and subsequently lysed using a reaction buffer containing 0.22
M sucrose, 0.12 M mannitol, 40 mM tricine (pH 7.5) and 1 mM EDTA.
ATP production was measured using an Optocomp I BG-1 luminometer
(GEM Biomedical, Inc.).
Measurement of telomere length and
mitochondrial DNA (mtDNA) copy number
Total DNA (including nuclear and mitochondrial
genomic DNA) was extracted using a TIANamp total DNA kit (Tiangen
Biotech Co., Ltd.) and directly employed as a template for PCR. The
mtDNA copy number was then assessed by qPCR using SYBRGreen master
mix (Thermo Fisher Scientific, Inc.) and the ABI7500 Real-Time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
following primer pairs were used for qPCR: 200 nM mitochondrially
encoded NADH:ubiquinone oxidoreductase core subunit 1 (ND1)
forward, 5'-CCCTAAAACCCGCCACATCT-3' and reverse,
5'-GAGCGATGGTGAGAGCTAAGGT-3'; and 167 nM cytoglobin (HGB) forward,
5'-GTGCACCTGACTCCTGAGGAGA-3' and reverse,
5'-CCTTGATACCAACCTG-CCCAG-3'. The following thermocycling
conditions were used for qPCR: Initial denaturation, 98˚C for 30
sec, followed by 40 cycles of 98˚C for 10 sec and 60˚C for 60 sec.
mtDNA copy numbers were normalized to the nuclear single-copy gene
HGB. The relative mtDNA copy number was calculated using the
2-ΔΔCq method (13).
Mitochondrial transcriptional
activity
Total RNA was extracted from cells using
TRIzol® reagent (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. Total RNA was reverse
transcribed into cDNA using the RT-for-PCR kit (Qiagen, Inc.) in
accordance with the manufacturer's protocol. Subsequently, qPCR was
performed using SYBRGreen master mix (Thermo Fisher Scientific,
Inc.) and the ABI7500 Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The following primer pairs were
used for qPCR: Mitochondrially encoded cytochrome c oxidase I
forward, 5'-ATGCGGCCATAGGTTCTGC-3' and reverse,
5'-TCCTCAAGATGTCTCAGTTCCAT-3'; ND1 forward,
5'-TCGTCATAATCTGTCCCTACACA-3' and reverse,
5'-CGGCTTCGGCTCTTAGCAAA-3'; β-actin forward,
5'-CATGTACGTTGCTATCCAGGC3' and reverse,
5'-CTCCTTAATGTCACGCACGAT-3'. The following thermocycling conditions
were used for qPCR: Initial denaturation at 98˚C for 30 sec,
followed by 45 cycles of 98˚C for 10 sec and 60˚C for 60 sec. mRNA
expression levels were normalized to the internal reference gene
β-actin. Relative expression levels were calculated using
2-ΔΔCq method (13).
Western blotting
Total protein was extracted using the SoniConvert™
system (DocSense) and RIPA buffer (Sigma-Aldrich; Merck KGaA).
Total protein was denatured at 100˚C for 10 min and was
quantitatively measured using a bicinchoninic acid assay kit
(Sigma-Aldrich; Merck KGaA) in accordance with the manufacturer's
protocol. Total protein (20 µg) were separated by 8-16% SDS-PAGE
and transferred to nitrocellulose membranes. After blocking in 5%
bovine serum albumin (Sigma-Aldrich; Merck KGaA) at room
temperature for 30 min, the membranes were incubated with primary
antibodies targeted against microtubule associated protein 1 light
chain 3B (LC3B; cat. no. ab48394; 1:1,000; Abcam), p62 (cat. no.
ab109012; 1:2,000; Abcam) and β-actin (cat. no. ab8226; 1:5,000;
Abcam) at room temperature for 1 h. After three washes with PBS-T,
membranes were incubated with horseradish peroxidase-conjugated
goat anti-rabbit secondary antibodies (cat. no. ab7090; 1:5,000;
Abcam) at room temperature for 1 h. Protein bands were visualized
using a chemiluminescence kit (Thermo Fisher Scientific, Inc.).
Protein expression levels were quantified using Image J software
(version 1.51; National Institutes of Health) with β-actin as the
loading control.
Statistical analysis
Data are presented as the mean ± standard error of
the mean and analyzed using SPSS software (version 16.0; SPSS,
Inc.) All experiments were repeated 3 times independently.
Comparisons among multiple groups were analyzed using one-way ANOVA
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
GA exerts protective effects against
H/R-induced cellular injury in HCAECs
To evaluate the effect of GA on H/R-induced cellular
injury in HCAECs, different concentrations of GA (0-200 µM) were
added to the medium during H/R induction, and the CCK-8 assay was
subsequently performed after 4, 8 or 12 h. The results suggested
that 100, 150 and 200 µM GA significantly decreased H/R-induced
cell injury at each time point compared with the 0 µM GA group
(Fig. 1A). At each time point, there
were no significant differences between the protective effects of
100 and 200 µM GA; therefore, 100 µM GA was used for subsequent
experiments.
Considering that H/R induces cell injury and
decreases cell viability mainly by inducing apoptosis (13), apoptotic cell death following GA
incubation was assessed at different time points. GA incubation
significantly decreased the Annexin
V-FITC+/PI- and Annexin
V-FITC+/PI+ subpopulations at each time point
compared with the H/R group (Fig.
1B). The highest proportion of apoptotic cells was observed at
4 h after H/R induction, indicating that H/R-induced cell injury
peaked at this time point.
GA decreases total and mitochondrial
ROS levels and consequently decreases H/R-induced apoptosis
Considering that H/R induces intracellular and
mitochondrial ROS accumulation (14-17),
whether GA altered H/R-induced ROS accumulation was investigated
and the rate of apoptosis was assessed. By detecting intracellular
ROS (with DCFH-DA labeling) and mitochondrial ROS (with MitoSOX™
Red), total ROS and mitochondrial ROS levels after GA treatment
were measured. H/R-induced ROS accumulation was significantly
decreased by incubation with 100 µM GA (Fig. 2A). In addition, H/R-induced
mitochondrial ROS accumulation was decreased by incubation with 100
µM GA, which exhibited similar effects compared with treatment with
1 µM MitoQ10 (Fig. 2B),
indicating that GA and MitoQ10 elicited similar
effects.
Subsequently, the effects of GA on cell viability
and apoptosis were measured. A CCK-8 assay was performed and the
results revealed that GA treatment significantly increased the
H/R-induced reduction of cell viability, which was an effect
similar to that of NAC and MitoQ10 treatments (Fig. 2C). Annexin-FITC/PI double staining
followed by flow cytometry was performed to detect apoptosis.
Treatment with GA, N-acetylcysteine (NAC) or MitoQ10
decreased H/R-induced cell apoptosis (Fig. 2D). The results indicated that
H/R-induced ROS accumulation was important for inducing apoptosis,
and that GA treatment potentially displayed a protective effect
against cell apoptosis by decreasing ROS accumulation.
GA treatment maintains the
mitochondrial membrane potential and mitochondrial function
To investigate the effects of H/R-induced ROS
accumulation on the mitochondrial membrane potential, JC-1 staining
was performed. The mitochondrial membrane potential was notably
decreased following H/R induction, as suggested by a decrease in
red fluorescence compared with the normoxia group (Fig. 3A). Treatment with 100 µM GA, 10 µM
NAC or 1 µM MitoQ10 markedly increased red fluorescence
compared with the H/R group, which indicated that the 3 treatments
maintained the mitochondrial membrane potential following H/R
induction. To further investigate mitochondrial function, ATP
synthesis, mtDNA copy number and mitochondrial transcriptional
activity were assessed (Fig. 3B-D).
Treatment with GA or NAC significantly protected cells against
H/R-induced mitochondrial dysfunction, decreasing ATP synthesis and
normalizing transcriptional activity to that of the internal
reference gene (β-actin). In addition, the relatively high mtDNA
copy number following GA treatment was normalized to HGB, which is
a nuclear single-copy gene. The results revealed that GA treatment
was potentially associated with promotion of mitochondria
biogenesis or inhibition of mitophagy.
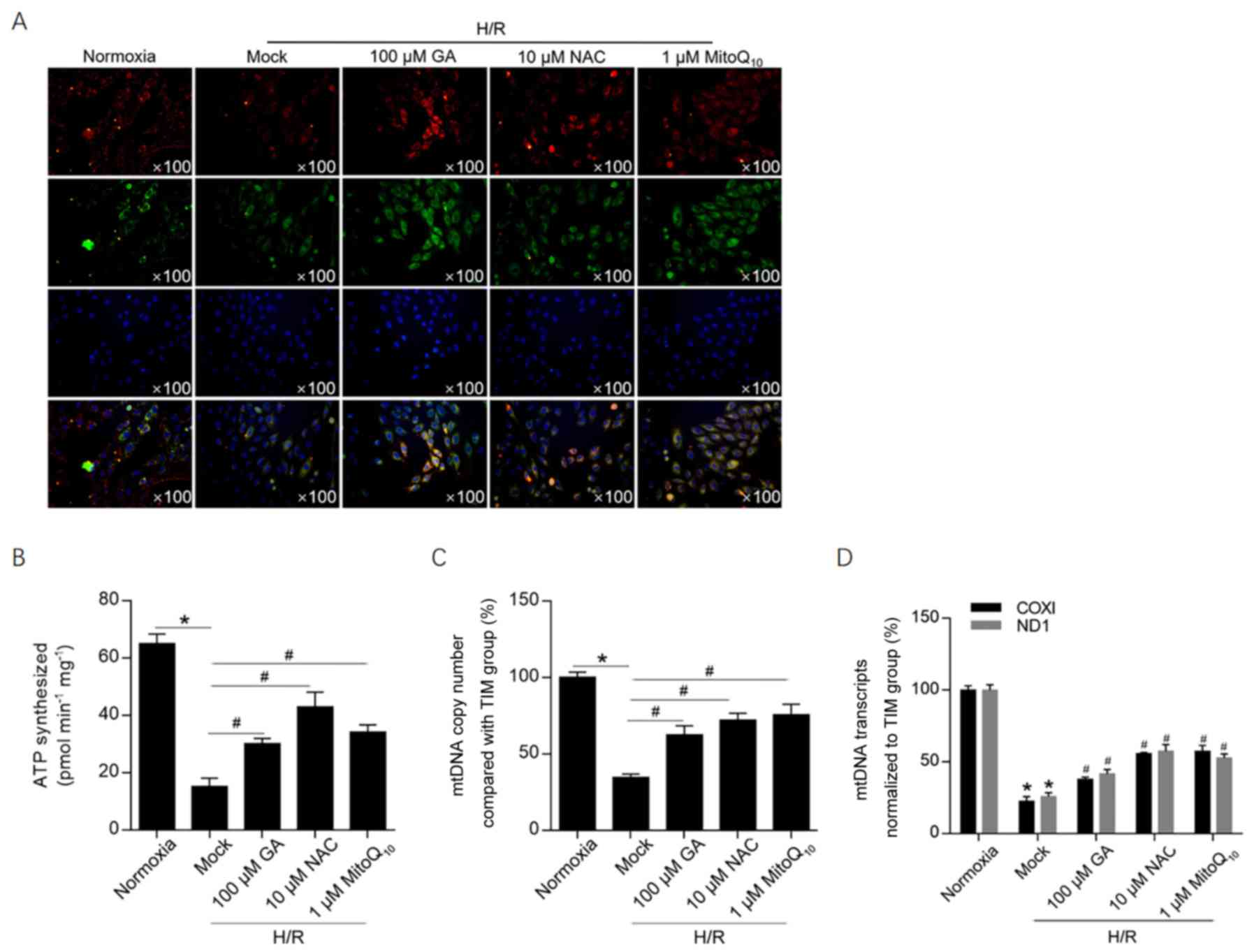 | Figure 3GA maintains the mitochondrial
membrane potential potentially via decreasing mitochondrial ROS
accumulation. (A) JC-1 staining was performed to investigate the
effect of GA on the mitochondrial membrane potential.
Multi-aggregates are stained red and mono-aggregates are stained
green. Following treatment with 100 µM GA, 10 µM NAC or 1 µM
MitoQ10, (B) ATP synthesis, (C) mtDNA copy number and (D) mtDNA
transcriptional activity were assessed. *P<0.05 vs.
normoxia group. #P<0.05 vs. H/R + mock group. GA,
glycyrrhizic acid; ROS, reactive oxygen species; NAC,
N-acetylcysteine; mtDNA, mitochondrial DNA; H/R,
hypoxia/reoxygenation; TIM, Rho guanine nucleotide exchange factor
5; COXI, mitochondrially encoded cytochrome c oxidase I; ND1,
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit
1. |
GA decreases autophagy/mitophagy and
autophagy induction abolishes its protective effects
To determine whether GA treatment maintained
mitochondrial function via autophagy/mitophagy, markers of
autophagy/mitophagy, including LC3B and p62, were detected by
western blotting. H/R-induced LC3B-II expression was significantly
decreased by GA, NAC or MitoQ10 treatment. Similarly,
the expression of p62, which is downregulated during
autophagy/mitophagy, was also markedly maintained by treatment with
GA, NAC or MitoQ10 following H/R induction (Fig. 4A).
To further clarify whether GA exerted protective
effects by inhibiting autophagy/mitophagy, cell viability and
apoptosis were analyzed in cells treated with GA and rapamycin, an
autophagy-inducing compound. Rapamycin exposure abolished
GA-induced protective effects on cell viability and apoptosis,
which indicated that the protective effects of GA were associated
with a decrease in autophagy/mitophagy (Fig. 4B and C).
Collectively, the results suggested that GA
decreased H/R-induced ROS accumulation, maintained the
mitochondrial membrane potential and inhibited mitophagy/autophagy,
which protected cells against H/R-induced injury.
Discussion
In the present study, the protective effects of GA
against H/R-induced decreases in cell viability and increases in
cell apoptosis in HCAECs were investigated. Treatment with GA
significantly inhibited H/R-induced ROS accumulation, maintained
the mitochondrial membrane potential and mitochondrial homeostasis,
and prevented mitochondrial dysfunction following H/R.
I/R and H/R are common causes of tissue and cell
damage due to a lack of blood supply or oxygen. Unfortunately,
restoration of the blood or oxygen supply leads to further
oxidative damage and inflammatory effects by inducing oxidative
stress (1). Certain endothelial
cells in different organs are particularly sensitive to ischemia
and hypoxia due to their direct contact with the circulation, and
damage to these cells can result from an oxidative stress imbalance
arising from the inflammatory response (18). I/R- and H/R-induced ROS accumulation
damages mitochondria, decreases the mitochondrial membrane
potential, damages DNA, lipids and proteins, and promotes mitophagy
(16). Considering the critical
effects of ROS in promoting cell injury following H/R, scavenging
ROS is a promising treatment strategy. N-n-butyl haloperidol
iodide (F2) has been reported to exert protective
effects against myocardial I/R injury by downregulating H/R-induced
ROS levels, inactivating JNK and downregulating early growth
response 1 protein expression in H9c2 cells (19). Metformin, a first-line drug for the
management of type 2 diabetes, has been reported to protect
cardiomyocytes against H/R injury by inhibiting ROS generation and
inflammatory responses (20).
Collectively, the aforementioned studies indicated that GA might
protect against H/R-induced injury by regulating ROS levels.
In the present study, H/R induced intracellular and
mitochondrial ROS accumulation, and treatment with GA significantly
decreased ROS levels. Further investigation is required to
determine whether ROS generation or accumulation is affected by GA.
To distinguish mitochondrial ROS from intracellular ROS, 0.5 µM
MitoSOX with an excitation wavelength of 365 nm was used. H/R
induced intracellular and mitochondrial ROS accumulation; however,
whether the observed increase in mitochondrial ROS levels was
amplified by intracellular ROS accumulation, as has been previously
reported, was not investigated in the present study (21,22).
Although H/R-induced intracellular ROS accumulation directly
interacts with mitochondrial proteins and lipids to accelerate
mitochondrial dysfunction (23), in
the present study, scavengers of intracellular and mitochondrial
ROS decreased the rate of apoptosis following H/R, suggesting that
mitochondrial ROS directly induced apoptosis. A decrease in the
percentage of Annexin V+/PI- and Annexin
V+/PI+ cells following treatment with GA in
H/R-induced cells was also observed, which suggested that necrosis
had also occurred; however, the effects of GA on necrosis require
further investigation.
An excess of mitochondrial ROS leads to mitophagy
and subsequent apoptosis, which is accompanied by a decrease in ATP
production and collapse of the mitochondrial membrane potential
(24,25). It has also been reported that GA
exerts a protective role against hypoxia-induced mitochondrial
damage by regulating mitochondria (26). Following H/R, GA treatment inhibited
the decrease in ATP production, the collapse of the mitochondrial
membrane potential and the decrease in mtDNA copy number. By
detecting the hallmarks of autophagy/mitophagy, it was further
revealed that GA treatment inhibited ROS-induced
autophagy/mitophagy and subsequent cell apoptosis by decreasing ROS
levels. The effects of GA on the viability of cells following H/R
induction was analyzed using the CCK-8 assay. Future studies should
perform time-lapse live-cell experiments to reveal the effects of
GA on myocardial cells and H/R-induced necrosis.
The present study had several limitations. Firstly,
H/R-induced mitochondrial ROS accumulation may induce endothelial
injury; however, whether the observed increase in mitochondrial ROS
levels was amplified by intracellular ROS was not investigated in
the present study. Secondly, the exact mechanism underlying
GA-induced reductions in ROS accumulation was not identified in the
present study and requires further investigation.
Acknowledgements
The authors would like to thank Mrs Yun Bai (Third
Military Medical University, Chongqing) for language editing the
manuscript.
Funding
The present study was supported by the Starting
Scientific Foundation of Zunyi Medical University (grant no.
SS20180601ZF).
Availability of data and materials
The datasets generated and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
QT designed the study. YC, WX and XK performed the
cellular experiments and analyzed the data. JZ and YX collected the
data, wrote the manuscript and performed the flow cytometry
experiments. DL contributed to designing the study and writing the
manuscript, and also provided supervision. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Turer AT and Hill JA: Pathogenesis of
myocardial ischemia-reperfusion injury and rationale for therapy.
Am J Cardiol. 106:360–368. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Fernandez-Jimenez R, Garcia-Prieto J,
Sanchez-Gonzalez J, Agüero J, López-Martín GJ, Galán-Arriola C,
Molina-Iracheta A, Doohan R, Fuster V and Ibáñez B: Pathophysiology
underlying the bimodal edema phenomenon after myocardial
ischemia/reperfusion. J Am Coll Cardiol. 66:816–828.
2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rezende PC, Ribas FF, Serrano CJ and Hueb
W: Clinical significance of chronic myocardial ischemia in coronary
artery disease patients. J Thorac Dis. 11:1005–1015.
2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bosetti F, Brizzi F, Barogi S, Mancuso M,
Siciliano G, Tendi EA, Murri L, Rapoport SI and Solaini G:
Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase)
activities in platelets and brain from patients with Alzheimer's
disease. Neurobiol Aging. 23:371–376. 2002.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Blackstone NW: The impact of mitochondrial
endosymbiosis on the evolution of calcium signaling. Cell Calcium.
57:133–139. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Saraste M: Oxidative phosphorylation at
the fin de siecle. Science. 283:1488–1493. 1999.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang C and Youle RJ: The role of
mitochondria in apoptosis*. Annu Rev Genet. 43:95–118.
2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cai X, Wang X, Li J and Chen S: Protective
effect of glycyrrhizin on myocardial ischemia/reperfusion
injury-induced oxidative stress, inducible nitric oxide synthase
and inflammatory reactions through high-mobility group box 1 and
mitogen-activated protein kinase expression. Exp Ther Med.
14:1219–1226. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Valls-Lacalle L, Barba I, Miro-Casas E,
Alburquerque-Béjar JJ, Ruiz-Meana M, Fuertes-Agudo M,
Rodríguez-Sinovas A and García-Dorado D: Succinate dehydrogenase
inhibition with malonate during reperfusion reduces infarct size by
preventing mitochondrial permeability transition. Cardiovasc Res.
109:374–384. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Xu M, Bi X, He X, Yu X, Zhao M and Zang W:
Inhibition of the mitochondrial unfolded protein response by
acetylcholine alleviated Hypoxia/Reoxygenation-Induced apoptosis of
endothelial cells. Cell Cycle. 15:1331–1343. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Guo X, Wu J, Du J, Ran J and Xu J:
Platelets of type 2 diabetic patients are characterized by high ATP
content and low mitochondrial membrane potential. Platelets.
20:588–593. 2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Saito T and Sadoshima J: Molecular
mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ
Res. 116:1477–1490. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Livak KD and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Cao X, Wang X, Ling Y, Song X, Yang P, Liu
Y, Liu L, Wang L, Guo J and Chen A: Comparison of the degree of
autophagy in neonatal rat cardiomyocytes and H9c2 cells exposed to
hypoxia/reoxygenation. Clin Lab. 60:809–814. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang Y, Shi G, Zheng J, Tang Z, Gao P, Lv
Y, Guo F and Jia Q: The protective effects of N-n-butyl haloperidol
iodide on myocardial ischemia-reperfusion injury in rats by
inhibiting Egr-1 overexpression. Cell Physiol Biochem. 20:639–648.
2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang Y, Chen G, Zhong S, Zheng F, Gao F,
Chen Y, Huang Z, Cai W, Li W, Liu X, et al: N-n-butyl haloperidol
iodide ameliorates cardiomyocytes hypoxia/reoxygenation injury by
extracellular calcium-dependent and -independent mechanisms. Oxid
Med Cell Longev. 2013(912310)2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Paradies G, Paradies V, Ruggiero FM and
Petrosillo G: Mitochondrial bioenergetics and cardiolipin
alterations in myocardial ischemia-reperfusion injury: Implications
for pharmacological cardioprotection. Am J Physiol Heart Circ
Physiol. 315:H1341–H1352. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Carden DL and Granger DN: Pathophysiology
of ischaemia-reperfusion injury. J Pathol. 190:255–266.
2000.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang Y, Liao H, Zhong S, Gao F, Chen Y,
Huang Z, Lu S, Sun T, Wang B, Li W, et al: Effect of N-n-butyl
haloperidol iodide on ROS/JNK/Egr-1 signaling in H9c2 cells after
hypoxia/reoxygenation. Sci Rep. 5(11809)2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hu M, Ye P, Liao H, Chen M and Yang F:
Metformin protects h9c2 cardiomyocytes from high-glucose and
hypoxia/reoxygenation injury via inhibition of reactive oxygen
species generation and inflammatory responses: Role of AMPK and
JNK. J Diabetes Res. 2016(2961954)2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Daiber A, Di Lisa F, Oelze M,
Kröller-Schön S, Steven S, Schulz E and Münzel T: Crosstalk of
mitochondria with NADPH oxidase via reactive oxygen and nitrogen
species signalling and its role for vascular function. Br J
Pharmacol. 174:1670–1689. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tajeddine N: How do reactive oxygen
species and calcium trigger mitochondrial membrane
permeabilisation? Biochim Biophys Acta. 1860:1079–1088.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liu P, Lin Y, Tang X, Zhang P, Liu B, Liu
Y and Miao F: Helix B surface peptide protects cardiomyocytes
against hypoxia/reoxygenation-induced apoptosis through
mitochondrial pathways. J Cardiovasc Pharmacol. 67:418–426.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ahn HJ, Kim KI, Kim G, Moon E, Yang SS and
Lee JS: Atmospheric-pressure plasma jet induces apoptosis involving
mitochondria via generation of free radicals. PLoS One.
6(e28154)2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Du JK, Cong BH, Yu Q, Wang H, Wang L, Wang
CN, Tang XL, Lu JQ, Zhu XY and Ni X: Upregulation of microRNA-22
contributes to myocardial ischemia-reperfusion injury by
interfering with the mitochondrial function. Free Radic Biol Med.
96:406–417. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Xu CL, Liang CH, Sun WX, Chen J and Chen
X: Glycyrrhizic acid ameliorates myocardial ischemic injury by the
regulation of inflammation and oxidative state. Drug Des Devel
Ther. 12:1311–1319. 2018.PubMed/NCBI View Article : Google Scholar
|

















