Introduction
Atherosclerosis is the main cause of the development
of multiple diseases, such as stroke, myocardial infarction and
peripheral arterial disease (1). The
dysregulation of lipid metabolism, notably the generation of
oxidized (ox) low-density lipoprotein (LDL), is an important
trigger for the development of atherosclerosis, causing vascular
endothelial dysfunction and induction of apoptosis (2). The vascular endothelium acts as a key
barrier of the vessel wall, resisting adhesion and migration of
inflammatory cells, which can promote the formation of the
atherogenic plaque (3). Plaque
rupture leads to the development of atherosclerotic-associated
complications, which are accompanied by high mortality (4). Therefore, the inhibition of endothelial
cell apoptosis is important to protect from infiltration of
inflammatory cells and from atherogenic plaque formation.
The endoplasmic reticulum (ER) is a key organelle
and is responsible for multiple lipid metabolism, whereas ER
dysfunction is considered a risk factor for chronic metabolic
diseases, including obesity, diabetes and insulin resistance
(3). Vascular endothelial cells
exhibit a high number of highly developed ER organelles, which
determine their sensitivity to ER dysfunction induced by
dyslipidemia and to the induction of endothelial cell apoptosis
(5). ER stress is used to correct
misfolded or unfolded proteins accumulating in ER, whereas it is
also produced if unfolded proteins are continuously overloaded.
Under these conditions, the balance and function of ER cannot be
re-established, resulting in a vicious cycle that damages cells and
leads to the induction of apoptosis and cytotoxicity (6). Therefore, the inhibition of ER stress
induction may be one of the potential ways to protect blood vessels
and inhibit atherosclerosis formation under conditions of abnormal
lipoprotein metabolism.
The ER stress sensors primarily include the protein
kinase RNA-like ER kinase (PERK) and the inositol-requiring protein
1α (IRE1α) which maintain their inactive state under normal
conditions by binding to the protein chaperone glucose-regulated
protein 78 (GRP78) in the ER lumen. However, following stimulation,
GRP78 is removed from these sensors and activates the ER
stress-associated signaling pathway (7). Endothelial cells stimulated by ox-LDL
indicate an enhancement in the levels of the C/EBPα-homologous
protein (CHOP) and caspase-12, which induce cell apoptosis
(8), indicating that the ER stress
levels have been abnormally elevated following ox-LDL stimulation.
A previous study used an atherogenic rabbit model and demonstrated
an increased expression of GRP78 in the endothelial layer of the
aorta, as well as a significant increase in the levels of CHOP
(9). The chronic glycolipid
metabolic abnormalities induced the accumulation of adipose tissue,
which was accompanied by high ER stress levels and was associated
with NLRP3 inflammasome activation leading to vascular endothelial
insulin resistance and exacerbating vascular damage (10). According to the aforementioned
studies, the application of drugs that can inhibit ER stress can be
used to restrain the formation of vascular injury-related
diseases.
Statins act mainly by inhibiting the cholesterol
synthetic pathway. It is hypothesized that statins may also have
pleiotropic effects, such as reduction of the inflammatory process
and esterification of cholesterol to macrophages (11). Therefore, the investigation of their
extensive mechanism of action could potentially increase the
efficacy of their therapeutic effects (12). Rosuvastatin has attracted
considerable attention in its clinical application due to the
significant reduction caused on LDL cholesterol levels compared
with the effects of atorvastatin and simvastatin at the same dose
(13). In addition, it remains
unclear whether the endothelial and cardiovascular benefits of
rosuvastatin are mediated by downregulating ER stress induced by
dyslipidemia at cell or tissue level, which is independent of
inhibiting cholesterol synthesis in atherosclerosis. Consequently,
the present study investigated the effects of rosuvastatin on
abnormal ER stress in human umbilical vascular endothelial cells
(HUVECs) stimulated with ox-LDL and in aortic tissues of
ApoE-/- mice in order to explore the protective effects
of this compound on endothelial cells.
Materials and methods
Chemical reagents
Human umbilical vein endothelial cells (HUVECs,
CL-0122) and endothelial cell culture medium were purchased from
Procell Life Science & Technology Co., Ltd. Rosuvastatin
(purity >98%; lot no. 511160104) was purchased from Lunan Better
Pharmaceutical Co., Ltd. Ox-LDL (YB-002) was purchased from
Guangzhou Yiyuan Biotechnologies Co., Ltd. Annexin V-FITC/
propidium iodide apoptosis detection kit (cat. no. C1062) and BCA
protein assay kit were supplied by Beyotime Institute of
Biotechnology. TRIzol® reagent was from Thermo Fisher
Scientific, Inc. TransScript Green Two-Step RT-qPCR SuperMix (cat.
no. AQ201-01) was from Beijing Transgen Biotech Co., Ltd.
Caspase-12 fluorometric assay kit was from BioVision, Inc. An
anti-eIF2α polyclonal antibody (cat. no. BS3651), an anti-GRP78
polyclonal antibody (cat. no. BS1154) and an anti-PERK polyclonal
antibody (cat. no. BS2156) were purchased from Bioworld Technology,
Inc. An anti-phosphorylated (p-)eIF2α (Ser 51) monoclonal antibody
(3398) and an anti-IRE1α monoclonal antibody (cat. no. 3294) were
from Cell Signaling Technology, Inc. Anti-p-PERK (cat. no. 40294)
polyclonal antibody and p-IRE1α (cat. no. 16927) polyclonal
antibody were obtained from Thermo Fisher Scientific, Inc. The
anti-GAPDH monoclonal antibody (cat. no. TA-08), HRP-conjugated
anti-rabbit IgG (cat. no. IH-0011) or anti-mouse IgG (cat. no.
IH-0031), as well as primers of CHOP, sXBP1 and
caspase-12 were from Beijing Dingguochangsheng Biotechnology
Co., Ltd. LDL assay kit (cat. no. A113-1), high density lipoprotein
(HDL) assay kit (cat. no. A112-1), total cholesterol (TC) assay kit
(cat. no. A111-1) and triglyceride (TG) assay kit (cat. no. A110-1)
were from Nanjing Jiancheng Bioengineering Institute.
Cell culture
HUVECs were cultured with endothelial cell culture
medium (Ham's F-12K) supplemented contain 10% fetal bovine serum
(FBS), 0.05 mg/ml endothelial cell growth supplement, 0.1 mg/ml
heparin and 1% penicillin/streptomycin at 37˚C and 5%
CO2.
Annexin V-FITC/ PI apoptosis
assay
HUVECs in the logarithmic growth phase were
dispersed by trypsinization, and seeded into 6-well plates at a
density of 1x105 cells/ml and 2 ml/well overnight at
37˚C. Subsequently, HUVECs pretreated with the indicated
concentration of rosuvastatin (0, 0.01, 0.1 and 1 µmol/l) (14) for 24 h respectively; then, the cells
were incubated with or without ox-LDL (200 µg/ml) for another 24 h
at 37˚C. Following treatment, HUVECs were dispersed by
trypsinization without any EDTA for 1 min and centrifuged at 1,000
x g for 5 min at 4˚C. Sedimentary cells were washed by pre-cooled
PBS three times and then resuspended in Annexin V-FITC combined
liquid, 5 µl Annexin V-FITC and 10 µl PI added, and incubated for
20 min in dark with ice bath. The cell apoptosis amounts were
detected with a flow cytometer (BD LSRFortessa, BD Biosciences)
within 30 min, the values were calculated by BD FACSDiva™ Software
(v.8.0, BD Biosciences, Inc.).
Reverse transcription-quantitative
(RT-q) PCR assay
HUVECs seeded into 6-well plates at a density of
1x105 cells/ml and 2 ml/well overnight at 37˚C, and
cells in the logarithmic growth phase were treated with the
indicated concentration of rosuvastatin and incubated with or
without ox-LDL. Firstly, HUVECs were harvested and lysed in 1 ml
TRIzol® reagent then mixed with 400 µl chloroform by
gently swirling. After resting for 5 min the mixture was
centrifuged at 12,000 x g for 15 min at 4˚C and 400 µl of the upper
aqueous phase collected. Isopropyl alcohol (400 µl) was added and
the mixture was centrifuged at 12,000 x g for 10 min at 4˚C. The
sedimentary RNA was washed with 75% ethanol, centrifuged at 12,000
x g for 5 min at 4˚C, resuspended in DEPC water and the OD value
detected at 260/280 nm (ratio 1.4-2.0). Subsequently, RNA was
reverse-transcribed with oligo (dT) primers, and qPCR conducted
with gene-specific primers in the presence of SYBR Premix Ex Taq
(Beijing Transgen Biotech Co., Ltd.), the total reaction volume was
20 µl. qPCR was conducted for three independent experiments, using
GAPDH as the housekeeping control. The RT-qPCR amplification
was performed with 40-60 cycles (95˚C, 5 sec; 55˚C, 15 sec; 72˚C,
10 sec) with the oligonucleotide primer sets as in Table I. The relative expression levels of
the target gene were calculated by the 2−ΔΔCq method
(15). All procedures were conducted
according to the manufacturer's protocol.
 | Table ISequence of amplified primers. |
Table I
Sequence of amplified primers.
| Primers | Forward | Reverse |
|---|
| CHOP
(NM_001195057.1) |
5'-GAACCAGGAAACGGAAACAG-3' |
5'-ATTCACCATTCGGTCAATCA-3' |
| sXBP1
(NM_005080.3) |
5'-GGATTCTGGCGGTATTGACT-3' |
5'-AGGGAGGCTGGTAAGGAACT-3' |
| Caspase-12
(NM_001191016.2) |
5'-CAGCACATTCCTGGTGTTTAT-3' |
5'-GACTCTGGCAGTTACGGTTGTT-3' |
| GAPDH
(NM_001289745.2) |
5'-AGAAGGCTGGGGCTCATTTG-3' |
5'-AGGGGCCATCCACAGTCTTC-3 |
Caspase-12 activity assay
HUVECs treated as previously described were
harvested with cell lysis buffer on ice for 10 min, the protein
concentration was determined with the BCA method and adjusted to
equal amounts of protein samples. Cell lysates (50 µl) were added
into 96 well plates, then 50 µl 2X reaction buffer containing 10
mmol/l DTT was added, as was 5 µl ATAD-AFC buffer. After incubation
for 1 h at 37˚C, the OD value was measured at 405 nm and the
relative activity of caspase-12 calculated. All samples were
measured according to the manufacturer's protocol using a Hitachi
7150 Biochemical Autoanalyzer (Hitachi, Ltd.).
Western blot analysis
HUVECs in the logarithmic growth phase were treated
with the indicated concentration of rosuvastatin, respectively, and
incubated with or without ox-LDL. Then HUVECs were harvested in
RIPA lysis buffer containing moderate protease inhibitor for 10 min
on ice, and the protein concentration determined with the BCA
method. The cell extract was centrifuged for 5 min at 14,000 x g
and 4˚C and equal amounts of protein samples (40 µg) loaded onto
8-12% polyacrylamide-SDS gels. After electrophoresis, the gels were
transferred to PVDF membranes which had been blocked with 5% (w/v)
skimmed milk for 1 h at room temperature. Subsequently, the
membranes were incubated with primary antibodies, including rabbit
anti-eIF2α polyclonal antibody (1:700), rabbit anti-p-eIF2α (Ser
51) monoclonal antibody (1:1,000), rabbit anti-GRP78 polyclonal
antibody (1: 700), rabbit anti-PERK polyclonal antibody (1:1,000),
rabbit anti-p-PERK polyclonal antibody (1:1,500), rabbit anti-IRE1α
monoclonal antibody (1:1,000), rabbit anti-p-IRE1α monoclonal
antibody (1:1,000) and mouse anti-GAPDH monoclonal antibody
(1:5,000) at 4˚C overnight. Finally, the bindings of target
proteins were detected with secondary antibody conjugated to HRP
(1:5,000) and visualized using ECL chemiluminescence (Beyotime
Institute of Biotechnology), then calculated the densitometry with
ImageJ software (version 1.51d; National Institutes of Health).
Atherosclerosis animal model
protocol
ApoE-/- male mice 20-22 g (n=16; 8 weeks
old) were obtained from Beijing Vital River Laboratory Animal
Technology and fed with atherogenic chow (a high-fat diet with 40
kcal% Fat, 1.25% Cholesterol), C57BL/6N male mice 20-22g (n=8; 8
weeks old) were purchased from Beijing Vital River Laboratory
Animal Technology and fed normal chow as normal control. After
atherogenic chow for 8 weeks, ApoE-/- mice were randomly divided
into two groups (n=8 each): Model group and rosuvastatin group (5
mg/kg). The rosuvastatin was dissolved in 0.5% carboxy methyl
cellulose sodium and given once a day by intragastric
administration; an equal volume of vehicle was given in the control
and model groups for 4 weeks. Then the mice were euthanized using
20 mg/ml pentobarbital sodium (120 mg/kg body weight) through the
intraperitoneal route, followed by cervical dislocation. During the
present study, the animals were housed in a temperature of 22‐26 ̊C
and a humidity of 50‐65% in a controlled environment with a 12-h
light/dark cycle. The mice had free access to water and food, the
padding was changed twice a week and the health status was observed
with no mortalities. For animal welfare considerations, the mice
were provided with tubular toys.
All the experiments in vivo were approved by
the Animal Experimental Ethical Inspection Committee of Jilin
University School of Pharmaceutical Sciences (ethical permission
code: 20190025).
Measurement of LDL, HDL, TC and TG
levels in serum
The mice were sacrificed following rosuvastatin
treatment and the blood plasma was collected to detect LDL, HDL, TC
and TG levels in serum. The methods of LDL and HDL detection were
similar: 2.5 µl serum from each group mice were added into 96 well
plates, 2.5 µl standard substance was also added as standard
control and distilled water was used as a blank control; 180 µl
surfactant were added into each well for 5 min at 37˚C and the OD
value measured at 546 nm. Then 60 µl surfactant 2 were added for 5
min at 37˚C, the OD value was measured again and the concentration
of LDL and HDL calculated. For the measurement of TC and TG, 2.5 µl
serum was mixed with 250 µl working fluid in 96-well plates for 10
min at 37˚C, with standard substance as standard control and
distilled water as a blank control; then the OD value was measured
and the concentration of TC and TG calculated. All the samples were
measured according to the manufacturer's protocol using a Hitachi
7150 Biochemical Autoanalyzer (Hitachi, Ltd.).
Histopathology and
immunohistochemistry assays
The whole aorta was completely removed, the adipose
tissue around the blood vessel peeled off, and the aorta dissected
longitudinally with precision scissors prior to being fixed with 4%
polyformaldehyde for 24 h at room temperature. Subsequently, the
aortas were washed with PBS and 60% isopropanol, and stained in oil
red O for 2 h in the dark at room temperature. Oil red O stained
the lipid-rich plaque red. The aortas were smoothed on a black
background and images captured; the total area and the red plaque
area was calculated using ImageJ software (version 1.51d; National
Institutes of Health).
The aortic root was dissected, removed, fixed in 4%
polyformaldehyde for 48 h at room temperature, embedded in paraffin
and cut into 5 µm-thick sections. The sections were stained using a
hematoxylin and eosin kit (Beyotime Institute of Biotechnology) for
plaque morphology at room temperature for 3 min. Other sections
were blocked in 5% bovine serum albumin (BSA) at room temperature
for 1 h and incubated with primary antibodies overnight at 4˚C,
then incubated with HRP-conjugated anti-rabbit IgG or anti-mouse
IgG (1:5,000 dilution). TUNEL (1:9 mixed in equilibration buffer)
staining of apoptotic cells was performed in the aorta for 1 h, and
depicted as green fluorescence. DAPI (5 µg/ml) dyed the nucleus for
5 min and showed blue fluorescence, and endomucin, the marker of
endothelial cells, showed red fluorescence. All stains were
performed at room temperature. Staining results were all observed
and images captured under the routine microscope and fluorescence
microscope, three fields per sample were observed, and analyzed
using Photoshop (v.13.0; Adobe Systems, Inc.) and Image-Pro Plus 6
6.0 (Media Cybernetics, Inc.) analysis software.
Statistical analysis
Statistical analysis was performed using the SPSS
v.22.0 statistical package (IBM Corp.). The results were expressed
as the mean ± standard deviation. Statistical differences among all
groups were evaluated using one-way analysis of variance with
Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
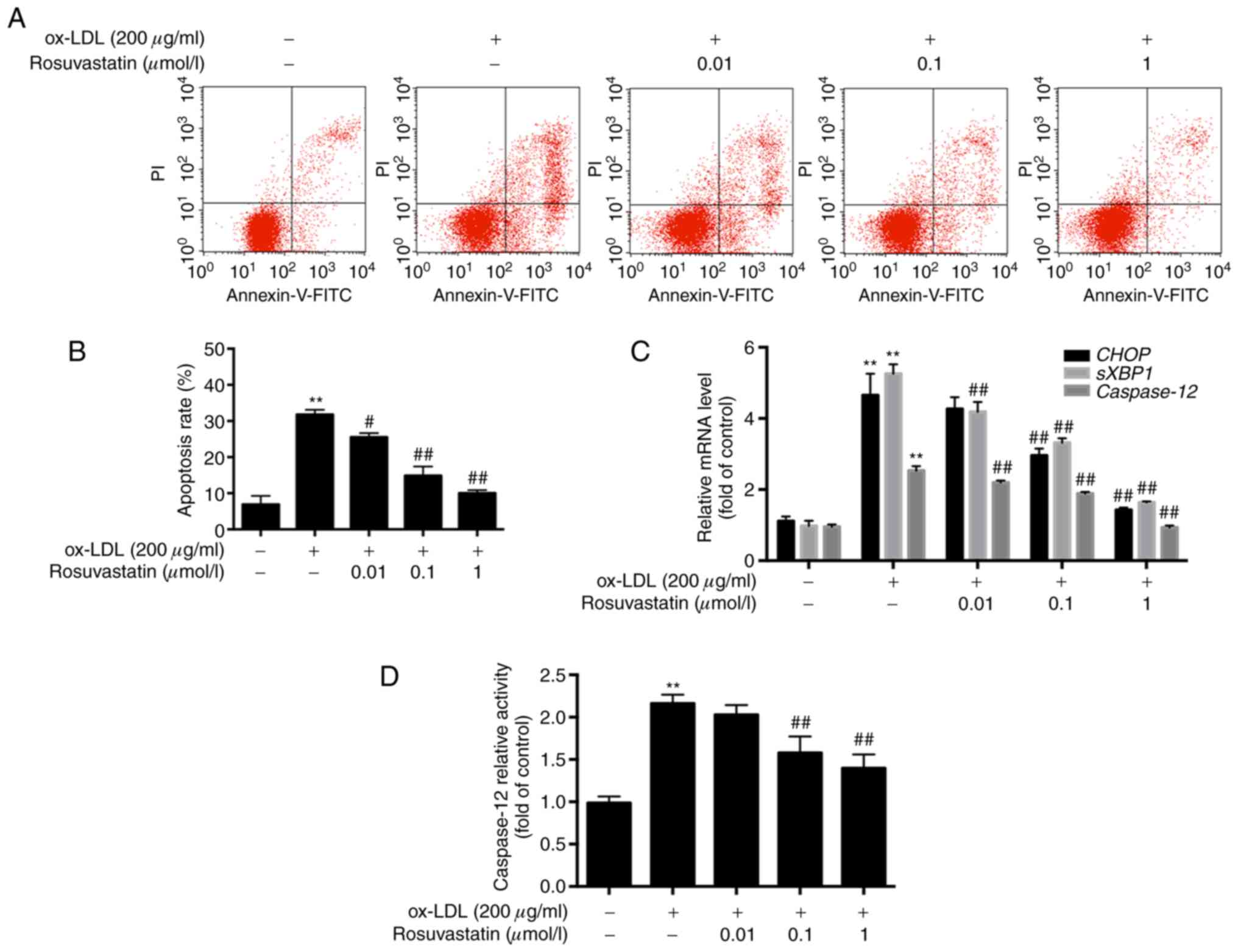
Effects of rosuvastatin on
ox-LDL-induced HUVEC apoptosis
HUVECs were pretreated with rosuvastatin and the
ox-LDL-mediated apoptotic rates were assessed by flow cytometry.
HUVECs treated with ox-LDL (200 µg/ml) exhibited increased
apoptosis. Specifically, the apoptotic rate of these cells was
increased to 30.78±2.74% compared with that of the control cells
(6.89±2.38%, P<0.01). However, the apoptotic rates of HUVECs,
which were pretreated with rosuvastatin (0.01, 0.1 and 1 µmol/l),
were decreased significantly in a concentration-dependent manner
(P<0.01). The results suggested that rosuvastatin reversed the
effects of ox-LDL-induced HUVEC apoptosis (Fig. 1A and B).
The results of the RT-qPCR analysis revealed that
the mRNA levels of CHOP, sXBP1 and caspase-12
were all significantly increased in ox-LDL-stimulated HUVECs
compared with those of the cells in the control group (P<0.01,
Fig. 1C), while HUVECs pretreated
with 0.1 and 1 µmol/l rosuvastatin exhibited lower mRNA levels of
CHOP compared with those of the ox-LDL stimulated HUVECs
(P<0.01). The mRNA levels of sXBP1 and caspase-12
in HUVECs pretreated with 0.01-1 µmol/l rosuvastatin were
significantly decreased in a concentration-dependent manner
(P<0.01). Rosuvastatin decreased the mRNA levels of ER
stress-associated apoptotic markers, suggesting that the inhibition
of HUVEC apoptosis may be associated with the repression of ER
stress hyperactivity. Furthermore, caspase-12 activity was
increased in ox-LDL simulated HUVECs compared with the control
(P<0.01, Fig. 1D), while 0.1 and
1 µmol/l rosuvastatin decreased caspase-12 activity in HUVECs
compared with the ox-LDL stimulated group (P<0.01).
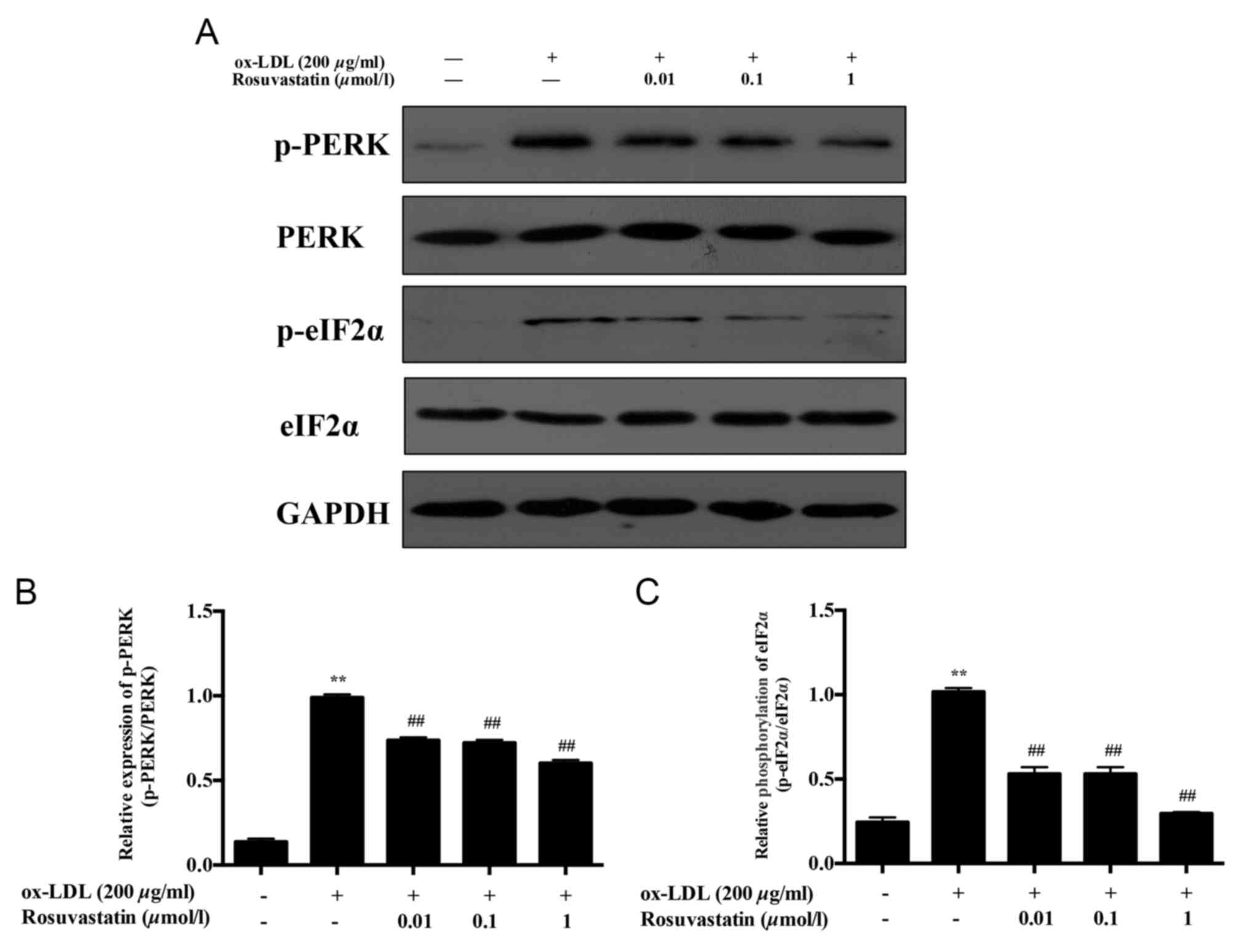
Effects of rosuvastatin on ER
stress-associated signaling pathways in HUVECs induced with
ox-LDL
The phosphorylation of PERK/eIF2α was proportional
to the increase noted in the mRNA levels of CHOP. The
phosphorylation levels of PERK and eIF2α were significantly
increased in ox-LDL HUVECs (Fig. 2,
P<0.01) compared with those in the ox-LDL stimulated group.
HUVECs pretreated with 0.01 and 0.1 µmol/l rosuvastatin resulted in
decreased expression levels of p-PERK (P<0.01), whereas 1 µmol/l
rosuvastatin treatment caused a more evident decrease in the
expression levels of p-PERK (P<0.01). The phosphorylation levels
of eIF2α in HUVECs pretreated with 0.01 and 0.1 µmol/l rosuvastatin
were decreased significantly (P<0.01). Treatment of the cells
with 1 µmol/l rosuvastatin decreased the phosphorylation of eIF2α
significantly (P<0.01), while the expression levels of total
PERK and eIF2α demonstrated no notable difference compared with
those of the control or ox-LDL induced HUVECs.
Concomitantly, the expression levels of ER-stress
sensor proteins, including GRP78 and p-IRE1α were investigated. The
levels of GRP78 and p-IRE1α in ox-LDL simulated HUVECs were
significantly increased compared with those of the control group
(Fig. 3, P<0.01). Following
treatment of the cells with rosuvastatin at a concentration range
of 0.01-1 µmol/l, the GRP78 and p-IRE1α levels were decreased in a
concentration-dependent manner (P<0.01). The results suggested
that rosuvastatin protected ox-LDL-induced HUVEC apoptosis by
repressing the ER stress.
Effects of rosuvastatin on
atherogenesis induced by high-fat diet in ApoE-/-
mice
Initially, the serum lipoprotein levels were
detected in each group of mice. Specifically, the serum LDL, TC and
TG levels of the model mice that were induced by high-fat diet were
significantly higher compared with those of the normal mice
(P<0.01; Table Ⅱ), whereas the HDL levels were significantly
reduced (P<0.01). Rosuvastatin treatment significantly changed
the levels of LDL and TG compared with those in the model group
(P<0.05), while the effects of rosuvastatin on HDL and TC were
not apparent.
The vascular intima of the normal mice was smooth
without atherosclerotic plaques, while a high number of
atherosclerotic plaques were noted in the aorta intima of
ApoE-/- mice fed with atherogenic chow for 12 weeks
(P<0.01). These data indicated that the atherosclerosis model
was successful and that rosuvastatin significantly alleviated
aortic plaque deposition compared with that of the model group
(P<0.01). In addition, the cross-section lesion areas were
significantly increased in the model group compared with those of
the normal group (P<0.01). The lesions were significantly
decreased in the rosuvastatin-treated group (P<0.01, Fig. 4A and B).
Effect of rosuvastatin on aortic
endothelial cell apoptosis
Endomucin is the marker of endothelial cells and is
stained with red fluorescence. A large amount of aortic endothelial
cell apoptosis was noted in atherosclerotic ApoE-/- mice
compared with normal mice (P<0.01; Fig. 5), whereas the induction of
endothelial cell apoptosis was significantly reduced following
treatment of the cells with rosuvastatin (P<0.01). These results
indicated that rosuvastatin could inhibit apoptosis of vascular
endothelial cells induced by high-fat diet.
Effect of rosuvastatin on ER stress in
the aortic intima of atherosclerotic mice
The expression levels of GRP78, p-PERK, p-IRE1α and
p-elF2α were investigated in the aortic intima of
ApoE-/- mice. GRP78, p-PERK, p-IRE1α and p-elF2α were
rarely expressed in the aorta intima of normal mice compared with
the strong expression which was noted in the intima and plaque of
model mice (P<0.01; Fig. 6).
Rosuvastatin inhibited the expression levels of the ER stress
signaling pathway proteins and decreased significantly the
expression levels of phosphorylated PERK, IRE1α and elF2α
(P<0.05). The results indicated that rosuvastatin could inhibit
ER stress in vascular endothelial cells treated with a high-fat
diet.
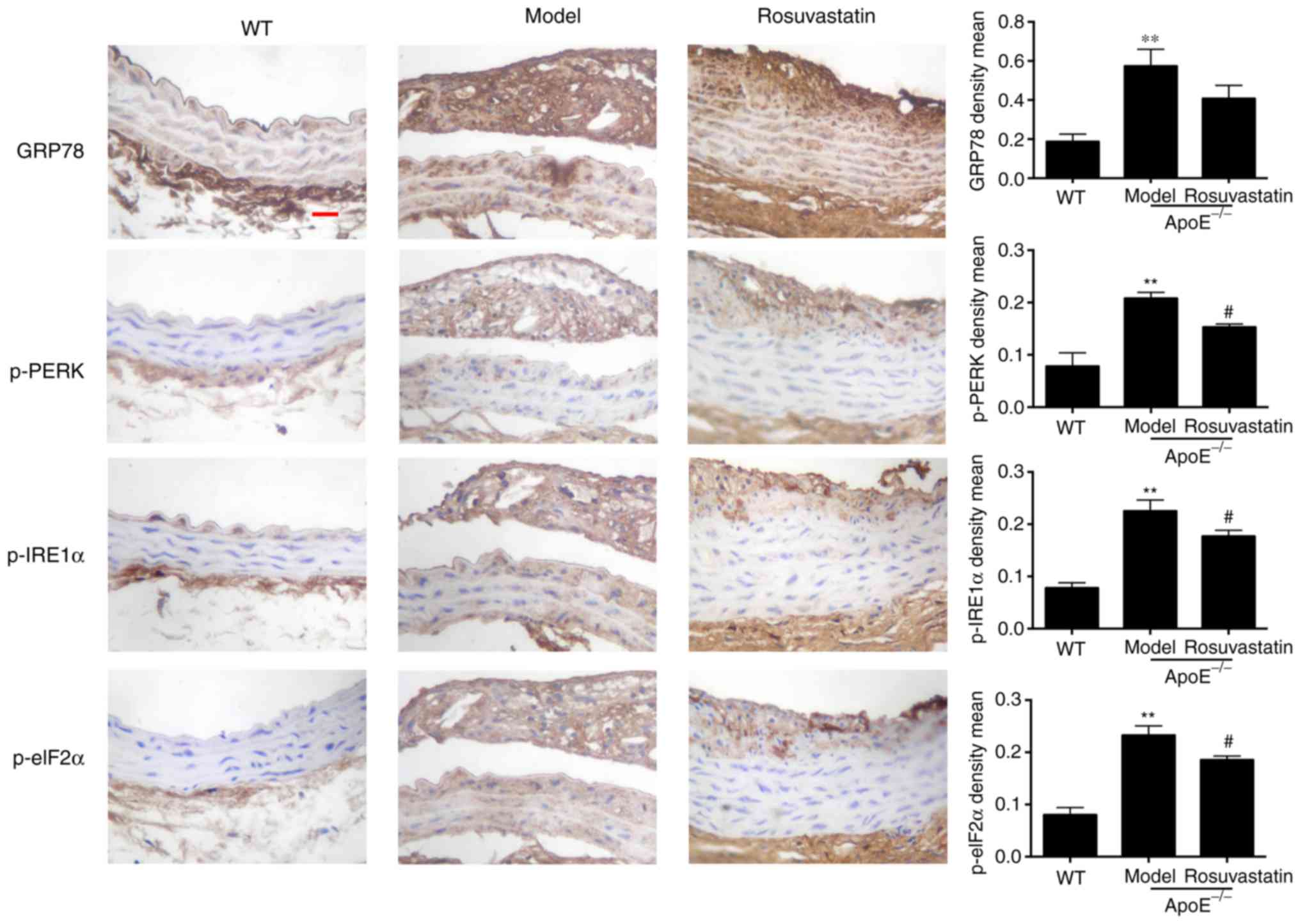 | Figure 6Effect of rosuvastatin on ER stress
in aortic intima of atherosclerotic mice. Representative
immunohistochemical staining and content quantification of GRP78,
p-PERK, p-IRE1α and p-elF2α in aortic intima and plaque,
magnification, x 400, scale bar = 50 µm. The density means
expressed as the mean ± standard deviation. **P<0.01
vs. Control group; #P<0.01 vs. Model group. ER GRP78,
glucose-regulated protein 78; p-, phosphorylated; PERK, protein
kinase RNA-like ER kinase; IRE1α, p-inositol-requiring protein 1α;
eIF2α, inositol-requiring protein 1α. |
Discussion
Atherosclerosis has attracted considerable attention
worldwide, due to its high risk of mortality in cardiovascular and
cerebrovascular diseases (16). The
disruption of lipid metabolism is critical for the formation of
atherosclerosis (17). Vascular
endothelial cells can limit the atherosclerotic process by
resisting inflammatory cell infiltration, which mainly results in
the formation of the atherosclerotic plaques (18). Dyslipidemia covers a wide range of
lipoprotein abnormalities, including abnormal levels of LDL, TC, TG
and HDL. LDL receptors are first upregulated in cholesterol
metabolism (19), and LDL ingested
and modified into ox-LDL exhibits a variety of proatherogenic
properties on cultured vascular cells (20). Thus the intervention of ox-LDL for
vascular cells in study of AS in vitro is widely used
(21). In the present study, HUVECs
were incubated with ox-LDL in vitro, which caused the
induction of apoptosis as determined by flow cytometry.
Furthermore, the cytotoxicity of ox-LDL and its ability to promote
endothelial cell apoptosis was significantly increased leading to
atherosclerotic plaque instability and rupture (22). The present study further indicated
that a high-fat diet led to significantly abnormal blood lipids and
severe atherosclerotic pathological changes in ApoE-/-
mice. These alterations included disorder in endothelial cell
arrangement and the formation of aortic plaques in the aorta
intima, as well as a large fraction of endothelial cell apoptosis.
The results suggested severe endothelial cell injury during
atherogenesis.
ER stress induced by ox-LDL is an important
mechanism of endothelial cell apoptosis that includes upregulation
of the PERK/eIF2α/CHOP signaling pathway, as well as the cleavage
of caspase-3. These processes are associated with ER stress-induced
apoptosis (23) and a variety of key
lipid synthesis pathways are located in the ER, which means
dyslipidemia can specifically stimulate abnormally high ER levels
associated with apoptosis (24). A
previous study indicated that ER functions are not only required
for the regulation of lipid metabolic disorders in the liver, but
they can also protect from endothelial cell homeostasis, since
increased levels of lipids can disturb ER homeostasis, leading to
ER stress and vascular injury (25).
Under the electron microscope, vascular endothelial cells can be
observed with highly developed ER, which suggests that endothelial
cells are more sensitive to ER (5,26). The
exposure of endothelial cells to ox-LDL induces a time-dependent
dissociation of PERK and GRP78 in human atherosclerotic lesions
that leads to splicing of IRE1α mRNA, which encodes for the
X-box-binding protein-1 (sXBP1) (24). In addition, upregulated mRNA levels
of caspase-12 are noted, which is the specific apoptotic
factor activated by ER stress (8).
The present study indicated that ox-LDL induced a considerable
increase in the mRNA levels of sXBP1 and caspase-12,
along with caspase-12 relative activity, indicating that ox-LDL
could induce ER stress-associated apoptosis. Notably, PERK
activated its specific downstream marker eIF2α, which played a key
role in upregulating the mRNA levels of CHOP, thereby
repressing ER stress-associated apoptosis in highly active
secretory endotheliocytes. This pathway could be considered an
important target for vascular protection (27). In the present study, the
ox-LDL-stimulated group demonstrated a significant increase in the
mRNA levels of CHOP and in the phosphorylation of eIF2α,
which was consistent with a previous study (8). Notably, the levels of the
characteristic protein of ER stress, GRP78, were significantly
increased, whereas PERK and IRE1α were highly phosphorylated. ER
stress has recently been identified as not only an important
mechanism of endothelial cell apoptosis, but also as a key risk
factor to plaque growth and instability in atherosclerosis
(22). The present study indicated
high expression levels of GRP78, as well as p-PERK, IRE1α and elF2α
in the aortic intimal plaque of atherosclerotic mice, indicating
that ER stress was overactivated. In addition, ER is the pivotal
storage site of intracellular Ca2+ and can accelerate
the biosynthesis of cholesterol in animals (28). Insufficient levels of Ca2+
induce abnormal protein folding in ER by inhibiting PCSK9
secretion, which can in turn degrade LDL receptor-dependent
cholesterol uptake in hepatocytes, thereby resulting in disturbed
lipid metabolism (29). Various
pathological conditions including homocysteinemia, hyperlipidemia,
high glucose levels and insulin resistance can lead to endothelial
dysfunction in part through the activation of ER stress (30). Previous studies have reported effects
of statins on homocysteine and high glucose-induced endothelial ER
stress at the cellular level (31,32).
However, statins can induce new onset diabetes mellitus, especially
in subjects prone to diabetes, which cannot be ignored (33). Therefore, the inhibition of
endothelial injury and apoptosis induced by ER stress is considered
a therapeutic strategy for the prevention of atherosclerosis.
Rosuvastatin is an inhibitor of HMG-CoA reductase,
which reduces cholesterol levels in the circulation by restricting
the generation of cholesterol precursors and stimulating catabolism
of LDL (26). This is achieved by
enhancing the expression of the LDL receptor in the hepatocyte
surface (34). By contrast, statins
exhibit multiple effects on atherosclerosis, rosuvastatin can
control the process of atherosclerotic cerebral infarction (ACI) in
patients, which is associated with inhibition of the expression of
OX40 ligand and the stimulation of the expression of PPAR-γ in
endothelial cells (35); however,
few studies have been performed on ER stress induced by
dyslipidemia in endothelial cells. To this end, it was hypothesized
that inhibition of ER stress may be another important protective
mechanism of rosuvastatin in aortic endothelium. Ox-LDL-stimulated
HUVECs were pretreated with 0.01-1 µmol/l rosuvastatin and the
induction of cell apoptotic rates indicated a marked decrease
compared with that noted in ox-LDL-treated cells. The mRNA levels
of CHOP, sXBP1 and caspase-12 in rosuvastatin
pretreated groups were notably suppressed following the increase in
rosuvastatin concentration and the caspase-12 activity was clearly
decreased. Notably, the expression levels of GRP78 and the
phosphorylation levels of PERK, IRE1α and eIF2α were all decreased
by rosuvastatin treatment in a concentration-dependent manner.
Furthermore, atherosclerotic mice were orally treated with
rosuvastatin and the incidence of the aortic plaque in aortic
intima was reduced. In addition, LDL and TG levels in the serum
were significantly reduced, along with the induction of apoptosis
in aortic endotheliocytes. The expression levels of the
phosphorylated proteins PERK, IRE1α and eIF2α were significantly
inhibited in the aorta intima, which verified the protective effect
of rosuvastatin on endothelium. A previous study demonstrated that
statins exert ‘pleiotropic’ therapeutic effects and can be used in
extensive clinical applications (36). The use of statins can protect against
liver cirrhosis and fibrosis by ameliorating the dysfunction of
hepatic endothelial cells via the upregulation of KLF-2 expression
(37), as well as the inhibition of
endothelial cell dysfunction, which is induced by chronic
intermittent hypoxia in obstructive sleep apnea patients. These
subjects did not exhibit comorbidities, such as high levels of LDL
and obesity, which could potentially increase coronary vascular
risk and stroke (38). For future
studies, it is intended to perform microarray detection and
bioinformatics analysis of mouse aorta to screen out which upstream
proteins, microRNA or long non-coding RNA may be involved in the
regulation of ER stress apoptosis by rosuvastatin, and further
verify the possible upstream factors in ox-LDL-induced endothelial
cells, which may contribute to the design of novel therapeutic
strategies that target the vascular endothelium for the prevention
and treatment of endothelial dysfunction-associated diseases.
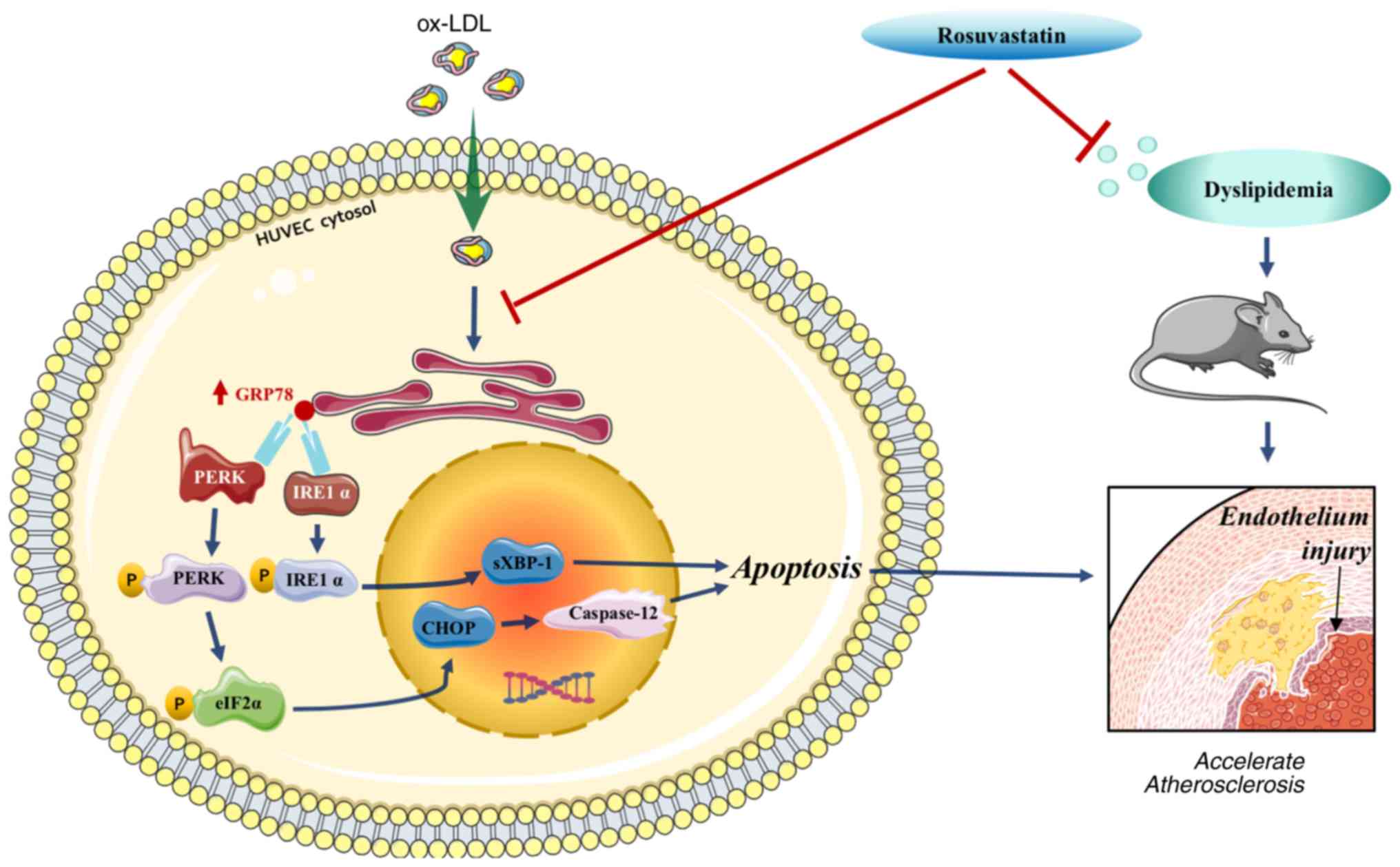
In conclusion, rosuvastatin reversed HUVEC
apoptosis, which was triggered by ox-LDL and endothelial injury
induced by a high-fat diet. These processes were mediated by
downregulating the PERK/p-eIF2α/CHOP and IRE1α/sXBP1 signaling
pathways (Fig. 7), indicating that
rosuvastatin may be conducive to enhance endothelial function. The
data suggested that rosuvastatin can be applied to the treatment of
other endothelial dysfunction-related diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Science and
Technology Development Projects of Jilin, China (grant no.
20150101200JC).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HX, DS and GL conceived and designed the study. JG,
HX, WF and XY performed the experiments. JG, DS and HX wrote the
paper. GX and HC analyzed the data. DS reviewed and edited the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All the animal experiments were approved by the
Animal Experimental Ethical Inspection Committee of Jilin
University School of Pharmaceutical Sciences (ethical permission
code: 20190025).
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hald EM, Lijfering WM, Mathiesen EB,
Johnsen SH, Løchen ML, Njølstad I, Wilsgaard T, Rosendaal FR,
Brækkan SK and Hansen JB: Carotid atherosclerosis predicts future
myocardial infarction but not venous thromboembolism: The Tromso
study. Arterioscler Thromb Vasc Biol. 34:226–230. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Byfield FJ, Tikku S, Rothblat GH, Gooch KJ
and Levitan I: OxLDL increases endothelial stiffness, force
generation, and network formation. J Lipid Res. 47:715–723.
2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Sarvani C, Sireesh D and Ramkumar KM:
Unraveling the role of ER stress inhibitors in the context of
metabolic diseases. Pharmacol Res. 119:412–421. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wu MY, Li CJ, Hou MF and Chu PY: New
insights into the role of inflammation in the pathogenesis of
atherosclerosis. Int J Mol Sci. 18(2034)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Luchetti F, Crinelli R, Cesarini E,
Canonico B, Guidi L, Zerbinati C, Di Sario G, Zamai L, Magnani M,
Papa S, et al: Endothelial cells, endoplasmic reticulum stress and
oxysterols. Redox Biol. 13:581–587. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Klausner RD and Sitia R: Protein
degradation in the endoplasmic reticulum. Cell. 62:611–614.
1990.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhou AX and Tabas I: The UPR in
atherosclerosis. Semin Immunopathol. 35:321–332. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hong D, Bai YP, Gao HC, Wang X, Li LF,
Zhang GG and Hu CP: Ox-LDL induces endothelial cell apoptosis via
the LOX-1-dependent endoplasmic reticulum stress pathway.
Atherosclerosis. 235:310–317. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kruzliak P, Sabo J and Zulli A:
Endothelial endoplasmic reticulum and nitrative stress in
endothelial dysfunction in the atherogenic rabbit model. Acta
Histochem. 117:762–766. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Xu X, Chen Y, Song J, Hou F, Ma X, Liu B
and Huang F: Mangiferin suppresses endoplasmic reticulum stress in
perivascular adipose tissue and prevents insulin resistance in the
endothelium. Eur J Nutr. 57:1563–1575. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Egom EE, Pharithi RB, Hesse S, Starr N,
Armstrong R, Sulaiman HM, Gazdikova K, Mozos I, Caprnda M, Kubatka
P, et al: Latest updates on lipid management. High Blood Press
Cardiovasc Prev. 26:85–100. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Breder I, Coope A, Arruda AP, Razolli D,
Milanski M, Dorighello GG, de Oliveira HC and Velloso LA: Reduction
of endoplasmic reticulum stress--a novel mechanism of action of
statins in the protection against atherosclerosis. Atherosclerosis.
212:30–31. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Karlson BW, Palmer MK, Nicholls SJ,
Lundman P and Barter PJ: A VOYAGER meta-analysis of the impact of
statin therapy on low-density lipoprotein cholesterol and
triglyceride levels in patients with hypertriglyceridemia. Am J
Cardiol. 117:1444–1448. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Geng J, Xu H, Yu X, Xu G, Cao H, Lin G and
Sui D: Rosuvastatin protects against oxidized low-density
lipoprotein-induced endothelial cell injury of atherosclerosis in
vitro. Mol Med Rep. 19:432–440. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tugcu A, Jin Z, Homma S, Elkind MS, Rundek
T, Yoshita M, DeCarli C, Nakanishi K, Shames S, Wright CB, et al:
Atherosclerotic plaques in the aortic arch and subclinical
cerebrovascular disease. Stroke. 47:2813–2819. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li D, Zhang L, Dong F, Liu Y, Li N, Li H,
Lei H, Hao F, Wang Y, Zhu Y, et al: Metabonomic changes associated
with atherosclerosis progression for LDLR(-/-) mice. J Proteome
Res. 14:2237–2254. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Rafieian-Kopaei M, Setorki M, Doudi M,
Baradaran A and Nasri H: Atherosclerosis: Process, indicators, risk
factors and new hopes. Int J Prev Med. 5:927–946. 2014.PubMed/NCBI
|
|
19
|
Goldstein JL and Brown MS: A century of
cholesterol and coronaries: From plaques to genes to statins. Cell.
161:161–172. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Goyal T, Mitra S, Khaidakov M, Wang X,
Singla S, Ding Z, Liu S and Mehta JL: Current concepts of the role
of oxidized LDL receptors in atherosclerosis. Curr Atheroscler Rep.
14:150–159. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhong X, Ma X, Zhang L, Li Y, Li Y and He
R: MIAT promotes proliferation and hinders apoptosis by modulating
miR-181b/STAT3 axis in ox-LDL-induced atherosclerosis cell models.
Biomed Pharmacother. 97:1078–1085. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Nègre-Salvayre A, Augé N, Camaré C,
Bacchetti T, Ferretti G and Salvayre R: Dual signaling evoked by
oxidized LDLs in vascular cells. Free Radic Biol Med. 106:118–133.
2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tao YK, Yu PL, Bai YP, Yan ST, Zhao SP and
Zhang GQ: Role of PERK/eIF2α/CHOP endoplasmic reticulum stress
pathway in oxidized low-density lipoprotein mediated induction of
endothelial apoptosis. Biomed Environ Sci. 29:868–876.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sanson M, Augé N, Vindis C, Muller C,
Bando Y, Thiers JC, Marachet MA, Zarkovic K, Sawa Y, Salvayre R, et
al: Oxidized low-density lipoproteins trigger endoplasmic reticulum
stress in vascular cells: Prevention by oxygen-regulated protein
150 expression. Circ Res. 104:328–336. 2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Röhrl C and Stangl H: Cholesterol
metabolism-physiological regulation and pathophysiological
deregulation by the endoplasmic reticulum. Wien Med Wochenschr.
168:280–285. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Dong Y, Fernandes C, Liu Y, Wu Y, Wu H,
Brophy ML, Deng L, Song K, Wen A, Wong S, et al: Role of
endoplasmic reticulum stress signalling in diabetic endothelial
dysfunction and atherosclerosis. Diab Vasc Dis Res. 14:14–23.
2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dickhout JG, Hossain GS, Pozza LM, Zhou J,
Lhoták S and Austin RC: Peroxynitrite causes endoplasmic reticulum
stress and apoptosis in human vascular endothelium: Implications in
atherogenesis. Arterioscler Thromb Vasc Biol. 25:2623–2629.
2005.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fu XL and Gao DS: Endoplasmic reticulum
proteins quality control and the unfolded protein response: The
regulative mechanism of organisms against stress injuries.
Biofactors. 40:569–585. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lebeau P, Al-Hashimi A, Sood S, Lhoták Š,
Yu P, Gyulay G, Paré G, Chen SR, Trigatti B, Prat A, et al:
Endoplasmic reticulum stress and Ca2+ depletion differentially
modulate the sterol-regulatory protein PCSK9 to control lipid
metabolism. J Biol Chem. 292:1510–1523. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lenna S, Han R and Trojanowska M:
Endoplasmic reticulum stress and endothelial dysfunction. IUBMB
Life. 66:530–537. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Jia F, Wu C, Chen Z and Lu G: Atorvastatin
inhibits homocysteine-induced endoplasmic reticulum stress through
activation of AMP-activated protein kinase. Cardiovasc Ther.
30:317–325. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhao SP, Wu ZH, Wu J, Hong SC and Deng P:
Effect of atorvastatin on tumor necrosis factor alpha serum
concentration and mRNA expression of adipose in
hypercholesterolemic rabbits. J Cardiovasc Pharmacol. 46:185–189.
2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chrysant SG: New onset diabetes mellitus
induced by statins: Current evidence. Postgrad Med. 129:430–435.
2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Raghow R: Statins redux: A re-assessment
of how statins lower plasma cholesterol. World J Diabetes.
8:230–234. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang JY, Liu B, Wang YN, Zhang WN and
Wang FJ: Effect of rosuvastatin on OX40L and PPAR-γ expression in
human umbilical vein endothelial cells and atherosclerotic cerebral
infarction patients. J Mol Neurosci. 52:261–268. 2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Adhyaru BB and Jacobson TA: Safety and
efficacy of statin therapy. Nat Rev Cardiol. 15:757–769.
2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Vargas JI, Arrese M, Shah VH and Arab JP:
Use of statins in patients with chronic liver disease and
cirrhosis: Current views and prospects. Curr Gastroenterol Rep.
19(43)2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Toraldo DM, Benedetto M, Conte L and De
Nuccio F: Statins may prevent atherosclerotic disease in OSA
patients without co-morbidities? Curr Vasc Pharmacol. 15:5–9.
2017.PubMed/NCBI View Article : Google Scholar
|