Introduction
Tumors deriving from chromaffin cells are rare
entities and can be divided into pheochromocytomas and
paragangliomas. Paraganglioma is a neuroendocrine tumor (NET)
originating in the paraganglia of the sympathetic and
parasympathetic nervous system. Paraganglia exhibit a widespread
distribution in the body, therefore paragangliomas can occur nearly
anywhere except within the bone and the brain (1). Mesenteric origin of these tumors is
highly unlikely and only a few cases have been reported in the
literature (2,3). We present a challenging case of an
unusual cystic tumor of the mesentery proved to be a
paraganglioma.
Case report
A 64-year-old female patient was admitted to the
hospital with symptoms of bowel obstruction and a palpable
abdominal mass. The patient was living in an urban environment and
did not have a significant prior medical history. Abdominal and
pelvic computed tomography revealed a cystic heterogeneously
enhanced mass measuring 80/66 mm in close proximity to the right
fallopian tubes, uterus and the small intestine. Furthermore, left
adrenal hyperplasia was observed on the computed tomography.
The tumor and the surrounding mesentery were
surgically removed as well as a segment of the corresponding small
intestine. Grossly, the tumor was located exclusively inside the
mesentery and it showed lesions of cystic degeneration and
hemorrhage (Fig. 1).
The histopathological examination was performed on
paraffin-embedded tissue samples which were further stained with
classical hematoxylin and eosin staining process.
Results
Microscopically, the mass was composed of nests of
small polygonal and round cells with central vesicular nuclei. Some
of them showed either eosinophilic or clear cytoplasm. The nests
were separated by connective tissue septa and the capsule of the
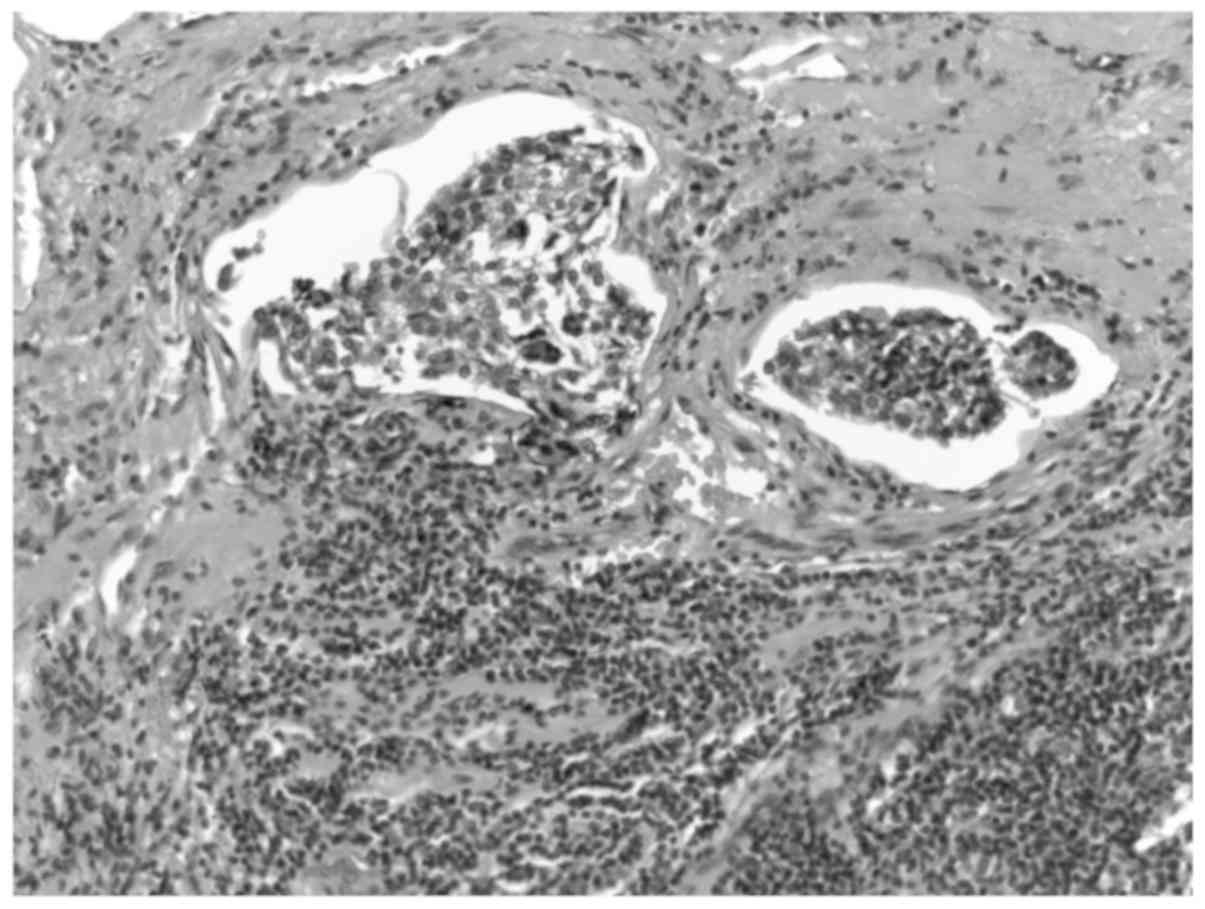
tumor was infiltrated by tumor cells (Fig. 2). Atypical mitoses were rarely
observed and necrosis was absent. Perineural and lymphovascular
invasion were clearly observed (Fig.
3). These microscopic findings were consistent with the
diagnosis of mesenteric paraganglioma.
Immunohistochemistry confirmed our diagnosis, as the
tumor cells were positive for Synaptophysin and Chromogranin A,
while S100 and glial fibrillary acidic protein (GFAP) markers
emphasized the presence of sustentacular cells. Antigen Ki-67
(Ki-67) index was under 1% (Fig. 4).
Extra markers were ordered for differential diagnosis, namely pan
cytokeratin (AE1/AE3), Desmin, clusted of differentiation 117
(CD117) and melanoma marker antibody (HMB45).
Finally, the histological grade of the tumor was
correlated with the risk of malignancy using the adrenal
pheochromocytoma and paraganglioma (GAPP) score. The score was
calculated using different parameters such as histological pattern,
score 1 (large and irregular cell nests); cellularity, score 2
(there were more than 250 cells/U*) [*U =
cells in unit of 10x10 µm under high power field (x400)]; comedo
necrosis, score 0 (the necrosis was absent); vascular or capsular
invasion, score 1 (both were present); Ki-67 labelling index, score
0 (Ki-67 <1%); catecholamine type, score 0 (there was no
documented production of catecholamines).
According to a total GAPP score of 4, the
paraganglioma was classified as moderately differentiated which
corresponds to an intermediate metastasizing risk.
Discussion
The mesentery is a rare location for extra-adrenal
paragangliomas. They are distributed along the para-aortic and
paravertebral axis corresponding to the sympathetic nervous system.
Abdominal occurrence is the best documented in the area
corresponding to the organ of Zuckerkandl (3,4).
The pathogenesis is not fully understood at the
moment, but as much as 40% of these tumors have been associated
with a germline mutation and therefore all patients with
paragangliomas should be referred for clinical genetic testing.
Hereditary cases may be associated with multiple endocrine
neoplasia type 2, von Hippel-Lindau disease, familial
paraganglioma, Carney triad and neurofibromatosis type 1(5). According to the European Clinical
Guideline, all patients diagnosed with pheochromocytoma and
paraganglioma should be followed-up for at least 10 years or should
be offered lifetime annual follow-up if criteria of high-risk were
present (young age, hereditary disease, large tumor and the
presence of a paraganglioma) (6).
The clinical presentation of paraganglioma resembles symptoms of
carcinoid syndrome, secondary to production of catecholamines.
These symptoms may include sweating, diarrhea, anxiety and
hypertension. However, it is not uncommon for a paraganglioma to
present itself without any symptoms, as it was shown in our
case.
Based on the 4th edition of WHO Endocrine Tumour
Classification (2017), all pheochromocytomas and paragangliomas
have a metastatic potential and they are all now considered
malignant, as opposed to a previous division into malignant and
benign tumors (7). However, the risk
of metastasis is correlated to the risk stratification based on
GAPP score which predicts the patient survival. GAPP score analyses
six parameters (histological pattern, cellularity, comedo necrosis,
vascular or capsular invasion, Ki-67 labelling index and
catecholamine type) and evaluates the malignant grade of
pheochromocytomas and paragangliomas. According to the total score,
the tumor falls into one of three categories (poorly, moderately
and highly differentiated) which are correlated to survival and
metastatic rate (8). In our case, a
score of 4 corresponds to a 60% metastatic rate and a 66.8% 5-year
survival (8).
Paragangliomas are microscopically described as
solid nests of round and oval cells with abundant granular
cytoplasm, which is either basophilic or acidophilic. These nests
of cells are designated as ‘Zellballen’ pattern which is highly
specific to paragangliomas. The nests are specifically surrounded
by sustentacular cells showing dendritic features which are
difficult to spot on classical hematoxylin-eosin stain, but can be
highlighted with S100 marker (1).
Melanin-like pigment might occur, in which case they are called
‘Pigmented paragangliomas’. The stroma presents a prominent
vascular network. In most cases the necrosis is absent and no
rosettes and acini are present. Unusual mitoses are rare.
Pathological diagnosis of paragangliomas is often
difficult to establish, especially in less common locations such as
mesentery or when there is a lack of symptoms secondary to
catecholamine excess. These tumors have a low incidence and
immunohistochemistry is a compulsory tool for a correct
differential diagnosis. The most important markers required for
this diagnosis are chromogranin A (or dopamine β-hydroxylase) and
S100. Chromogranin A is a specific neuroendocrine marker which is
positive in all functioning and non-functioning paragangliomas,
but, if negative, a diagnosis of paraganglioma should be excluded.
Similarly, dopamine β-hydroxylase marker is also decisive for this
diagnosis. S100 yields evidence for sustentacular cells. Other
neuroendocrine markers such as CD56 and synaptophysin are also
useful but not exclusive (8). Ki-67
proliferation index is required for the calculation of GAPP
score.
Due to a lack of specific histological features such
as zellballen pattern, we carefully approached the process of
differential diagnosis. We considered several entities that could
mimic this tumor. Firstly, reaction to keratin stains was analysed.
Only a few rare paragangliomas such as gangliocytic and cauda
equine-types were described in the literature as being positive to
keratin stains and negativity to these markers is highly suggestive
for paraganglioma (9). Therefore,
AE1/AE3 marker was performed and a negative result excluded a
neuroendocrine tumor, as all neuroendocrine tumors are usually
positive to this stain. Similarly, negative reaction for CD117 and
desmin ruled out a possible gastrointestinal stromal tumor.
Metastasis of melanoma was also ruled out by a negative response
for HMB45.
In conclusion, non-functioning paragangliomas
situated in unusual locations such as the mesentery are
challenging. In case of scarce specific features on classic stains,
a careful approach in differential diagnosis should be considered.
The critical markers for paragangliomas, chromogranin A and S100,
should be used first in the process of diagnosis, followed by other
valuable immunohistochemical markers to rule out other entities. A
long-term follow-up is extremely important following the diagnosis
of paraganglioma as all these tumors present malignant
potential.
Acknowledgements
Not applicable.
Funding
Not funding was received.
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors's contributions
AI performed the tumor histological examination and
was a major contributor in the writing of the manuscript. IAU
performed the immunohistochemistry examination of the tumor and was
a major contributor in the writing of the manuscript. AP analyzed
and interpreted the patient data regarding the hereditary
involvement. MMC analyzed and interpreted the patient data
regarding the neuroendocrine disease. RCP performed the radiography
interpretation of the tumor and was a major contributor in the
writing of the manuscript. FS analyzed and interpreted the genetic
substrate of the disease. TC analyzed and interpreted the patient
data regarding the surgery approach and was a major contributor in
the writing of the manuscript. MCD analyzed and interpreted the
patient data regarding the differential diagnosis of the patient
and was a major contributor in the writing of the manuscript. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient provided written informed consent for
the patient information to be published.
Competing interests
Not applicable.
References
|
1
|
Asa S, Ezzat S and Mete O: The diagnosis
and clinical significance of paragangliomas in unusual locations. J
Clin Med. 7(280)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kudoh A, Tokuhisa Y, Morita K, Hiraki S,
Fukuda S, Eguchi N and Iwata T: Mesenteric paraganglioma: Report of
a case. Surg Today. 35:594–597. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jaffer S and Harpaz N: Mesenteric
paraganglioma: A case report and review of the literature. Arch
Pathol Lab Med. 126:362–364. 2002.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lack EE, Cubilla AL, Woodruff JM and
Lieberman PH: Extra-adrenal paragangliomas of the retroperitoneum:
A clinicopathologic study of 12 tumors. Am J Surg Pathol.
4:109–120. 1980.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fujita T, Kamiya K, Takahashi Y, Miyazaki
S, Iino I, Kikuchi H, Hiramatsu Y, Ohta M, Baba S and Konno H:
Mesenteric paraganglioma: Report of a case. World J Gastrointest
Surg. 5:62–67. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Plouin PF, Amar L, Dekkers OM, Fassnacht
M, Gimenez-Roqueplo AP, Lenders JW, Lussey-Lepoutre C and Steichen
O: Guideline Working Group. European Society of Endocrinology
Clinical Practice Guideline for long-term follow-up of patients
operated on for a phaeochromocytoma or a paraganglioma. Eur J
Endocrinol. 174(G1-G10)2016. View Article : Google Scholar
|
|
7
|
Lloyd RV, Osamura YR, Kloppel G and Rosai
J: WHO classification of tumours of endocrine organs. WHO Press.
31:1770–1786. 2017.
|
|
8
|
Kimura N, Takekoshi K and Naruse M: Risk
stratification on pheochromocytoma and paraganglioma from
laboratory and clinical medicine. J Clin Med.
7(E242)2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Singh S, Asa SL, Dey C, Kennecke H,
Laidley D, Law C, Asmis T, Chan D, Ezzat S, Goodwin R, et al:
Diagnosis and management of gastrointestinal neuroendocrine tumors:
An evidence-based Canadian consensus. Cancer Treat Rev. 47:32–45.
2016.PubMed/NCBI View Article : Google Scholar
|