Introduction
The inflammatory response is the key pathogenesis of
the most common forms of heart disease and its processes underlie
various conditions related with injury of the cardiac muscle, such
as cardiomyopathy, myocardial infarction, sepsis and heart failure
(1,2). Lipopolysaccharide (LPS) is a major
component of the bacterial outer membrane, and plays a crucial role
in the initiation of several diseases (3). Numerous previous studies demonstrated
that LPS contributes to inflammation and apoptosis; for example,
LPS-induced acute respiratory distress syndrome, systemic
inflammation induced by a low dose of LPS in mice and
LPS-stimulated acute kidney injury (4,5). LPS can
also induce inflammation and apoptosis in cardiomyocytes (6).
LPS, as a stimulus, contributes to pro-inflammatory
responses, in addition to the increased expression of numerous
inflammatory cytokines, including tumor necrosis factor-α (TNF-α),
monocyte chemo-attractant protein (MCP)-1 and intercellular
adhesion molecule (ICAM)-1 in the heart (7). A previous study has demonstrated that
TNF-α shows direct negative inotropic effects and several features
of heart failure (HF). In HF, inflammation might be associated with
dysregulation of the TNF-α feedback system (8). A previous study identified that cardiac
inflammation is accompanied by overexpression of ICAM-1 and
vascular cell adhesion molecule (VCAM)-1(9). During the development of inflammation,
cellular adhesion molecules (CAMs) mediate the transendothelial
migration of immune-cells into the cardiac tissue. Those
infiltrated cells, as well as cardiomyocytes, produce
pro-inflammatory cytokines, such as TNF-α, interleukin (IL)-1β and
IL-18. These cytokines stimulate the expression of CAMs in a
positive feedback system. Furthermore, they have direct and
indirect detrimental effects on the heart (10).
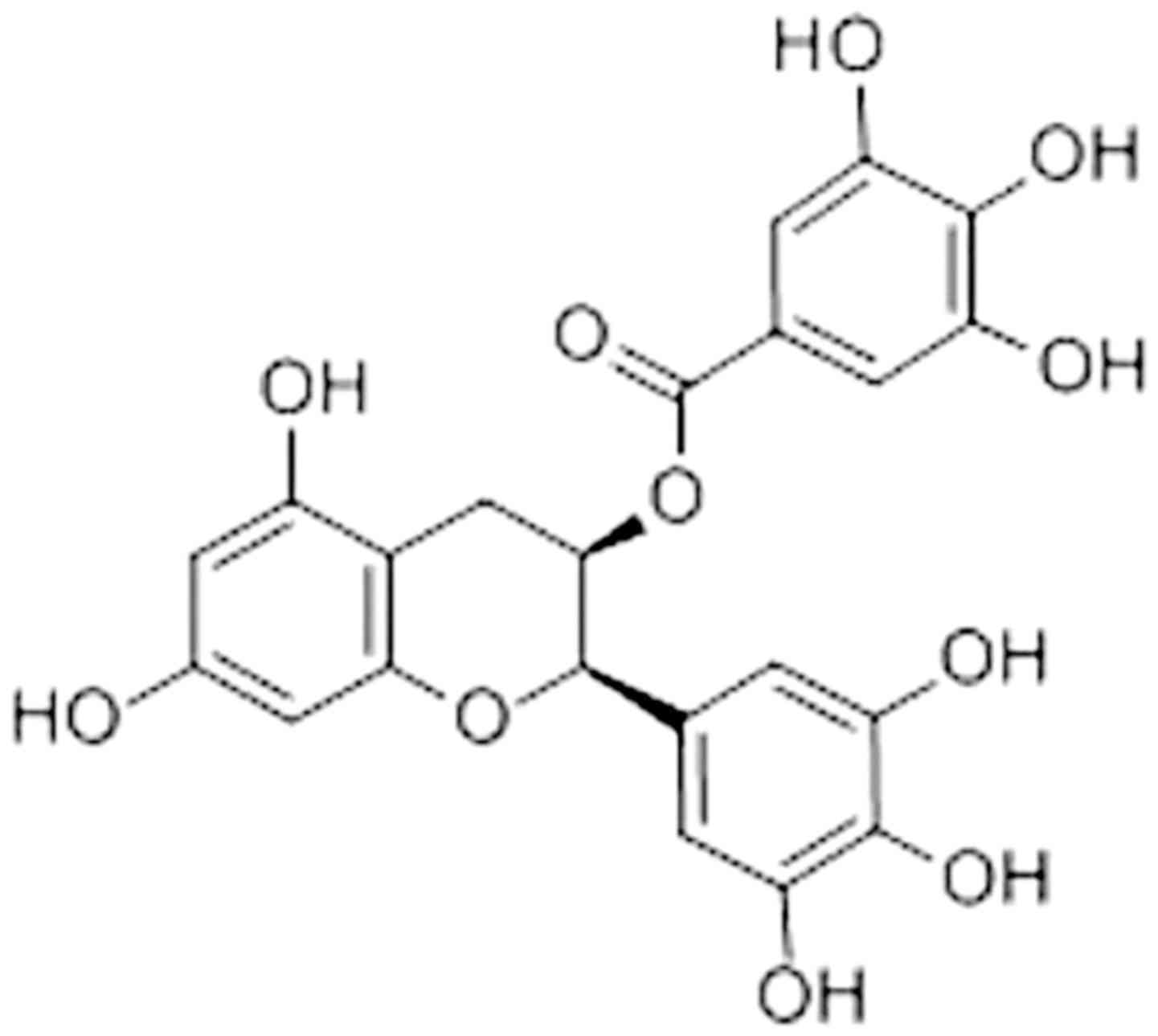
Epigallocatechin-3-gallate [EGCG; Fig. 1; (11)] is the major polyphenol extracted from
green tea, which is the most abundant and well-studied catechin
(12). Previous studies demonstrated
the beneficial effects of EGCG, including potential anti-oxidative,
anti-inflammatory and anti-tumorigenesis properties in the
treatment and prevention of several chronic diseases, including
heart diseases, cancer, obesity and endocrine disorders (13,14).
Among these effects, the anti-inflammatory activity of EGCG plays a
vital role against these diseases.
EGCG was demonstrated to decrease expression of
inflammatory genes, such as TNF-α, IL-1β, IL-6 and IL-8, when EGCG
(10 µg/ml) was applied to inflamed human corneal epithelial cells
(15). Similarly, there was a
significant downregulation of the expression of some kidney injury
markers and pro-inflammatory mediators in unilateral ureteral
obstruction (16). Previous studies
demonstrated that cardiac injuries were associated with the
activation of p38, PI3K-Akt and ERK 1/2(17). The PI3K-Akt pathway plays a vital
role in many biological reactions, including inflammatory
responses, chemotaxis, cellular activation and apoptosis, which
also regulates the expressions of inflammatory genes (18). Previous studies have demonstrated
that inflammation stimulates the activation of PI3K/Akt signaling
pathway in cardiomyocytes (19,20). A
previous study demonstrated that EGCG repressed the PI3K/Akt system
in fibroblast cells and phosphorylation of ERK and Akt kinases in
epidermal growth factor stimulated cells (21). However, to the best of the authors'
knowledge, few studies have investigated the effect of EGCG on
LPS-induced inflammation in H9c2 cells.
Therefore, the aim of the present study was to
investigate the regulation of EGCG on inflammatory mediators, and
examine whether EGCG treatment could ameliorate inflammatory
responses induced by LPS in H9c2 and the underlying mechanisms
in vitro.
Materials and methods
Reagents
EGCG (purity, 98%) was purchased from Sigma-Aldrich
(Merck KGaA). LPS was obtained from Sigma-Aldrich (Merck KGaA). The
Cell Titer 96 Aqueous cell viability assay kit was procured from
Promega Corporation. ELISA kits for vascular endothelial growth
factor (VEGF; cat. no. DY493), Rantes (cat. no. DY478), MCP-1 (cat.
no. SMJE00B), ICAM-1 (cat. no. MIC100), matrix metalloproteinase 2
(MMP-2; cat. no. SMMP200) and TNF-α (cat. no. SMTA00B), as well as
nitric oxide (NO; cat. no. SKGE001) assay kits were purchased from
R&D Systems, Inc. Anti-Akt (cat. no. 4685), anti-nuclear
factor-κB (NF-κB) p65 (cat. no. 3034), anti-p38 (cat. no. 9212),
anti-Erk (cat. no. 4695), anti-phosphorylated (p)-Akt (cat. no.
4060), anti-p-NF-κB p65 (cat. no. 3033) and anti-p-ERK (cat. no.
4376) were all obtained from Cell Signaling Technology, Inc. Mouse
anti-β-actin (cat. no. sc-58673) was obtained from Santa Cruz
Biotechnology, Inc.
Cell culture
The rat embryonic-heart derived cell line H9c2 was
obtained from The American Type Culture Collection. Cells were
cultured in DMEM (Sigma-Aldrich; Merck KGaA) supplemented with 10%
heat-inactivated FBS (Gibco; Thermo Fisher Scientific, Inc.), 25 mM
D-glucose, 100 U/ml penicillin and 100 U/ml streptomycin, and
maintained in a humidified atmosphere of 5% CO2 at 37˚C.
The medium was changed every 2 days.
Viability evaluation
Cell viability was determined by an MTS assay
(Promega Corporation). Cultured H9c2 cells (1x104
cells/well in 96-well plate) were treated with EGCG (0, 1.5, 3.0,
6.25, 12.5, 25, 50, 100 and 200 µmol/l) for 48 h, and 20 µl MTS
solution was added to each well. The cells were incubated for 1 h
at 37˚C in 5% CO2 and the absorbance was measured at 490
nm. The cell survival rate was determined using the optical density
(OD) as follows:
Cell survival rate
(%)=(ODtreated-ODblank)/(ODcontrol-ODblank)
x100.
ELISA analysis
Cultured H9c2 cells were treated with LPS (250
ng/ml) or LPS (250 ng/ml) + EGCG (0, 1.5, 3.0, 6.25, 12.5, 25, 50
and 100 µmol/l) for 24 h. Cultured supernatants were collected and
analyzed for the release of cyto- and chemokines using commercial
ELISA test systems. Levels of VEGF, Rantes, MCP-1, ICAM-1, MMP-2
and TNF-α were determined using ELISA kits (R&D Systems, Inc.).
Absorbance was measured at 450 nm, with the correction wavelength
set at 540 or 570 nm.
NO assay
Cultured H9c2 cells were treated with LPS (250
ng/ml) or LPS (250 ng/ml) + EGCG (0, 1.5, 3.0, 6.25, 12.5, 25, 50
and 100 µmol/l) for 24 h. Nitrite determination was detected using
Griess reagent. The absorbance was measured at 540 nm using a
flow-through spectrophotometer. The sensitivity of the NO assay was
<0.78 µmol/l.
Western blot analysis
Cultured H9c2 cells (5x106/10 cm dish)
were treated with LPS (250 ng/ml) or LPS (250 ng/ml) + EGCG (0,
1.5, 3.0, 6.25, 12.5, 25, 50 and 100 µmol/l) for 24 h. After
treatment, cells were washed twice with cold PBS and lysed with
RIPA lysis buffer (Beyotime Institute of Biotechnology) for 30 min
at 4˚C. Extracted protein in each cell lysate was determined using
a BCA protein assay kit (Pierce; Thermo Fisher Scientific, Inc.).
Equal amounts (20 µg) of protein were separated by SDS-PAGE on 10%
gels. Proteins were transferred to a PVDF membrane and blocked with
5% non-fat dry milk in PBS with 0.02% v/v Tween-20 for 1 h at room
temperature. The membrane was incubated for 16 h with primary
antibodies (all 1:1,000) in PBS-Tween at 4˚C. The membrane was
washed and incubated for 1 h at room temperature with a
peroxidase-labeled secondary antibody in PBS-Tween (anti-mouse,
cat. no. P0447; anti-rabbit, cat. no. P0448; Dako; Agilent
Technologies, Inc.). After further washing, the mumembrane was
detected with ECL chemiluminescence (Pierce; Thermo Fisher
Scientific, Inc.). ImageQuantTL software (LAS 4000; GE Healthcare)
was used for densitometry.
Statistical analysis
All experiments were performed in triplicate. The
data are presented as the mean ± SEM. One-way ANOVA and Tukey's
test were used to determine the statistical significance of
differences among the experimental groups and the control group.
SPSS 20.0 (IBM Corp) was used for statistical analyses. P<0.05
was considered to indicate a statistically significant
difference.
Results
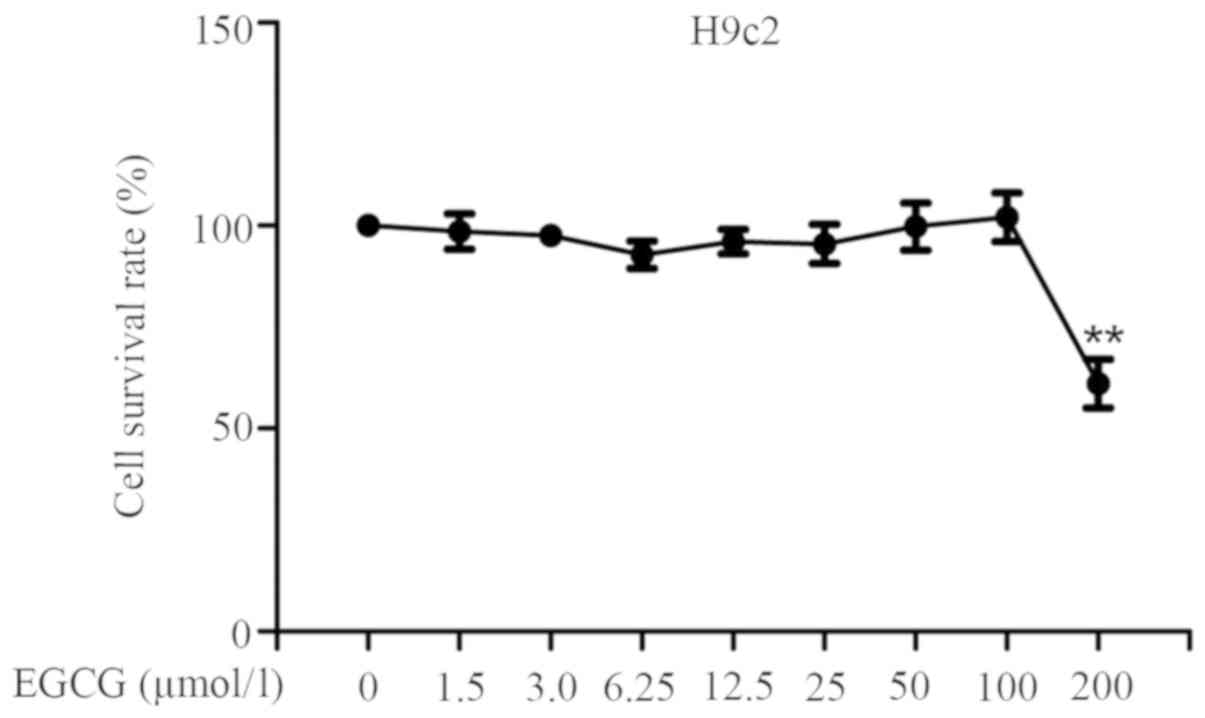
EGCG does not markedly affect cell
viability in H9c2 cells
The effects of EGCG on cell viability in H9c2 were
examined by the MTS assay. Cultured H9c2 cells (1x104
cells/well in a 96-well plate) were incubated without or with EGCG
(1.5, 3.0, 6.25, 12.5, 25, 50, 100 and 200 µmol/l) for 48 h. Cell
viability did not change significantly with respect to the control
up to 100 µM. However, 200 µM EGCG significantly reduced cell
viability and caused cytotoxicity in H9c2 cells (Fig. 2). Therefore, concentrations <200
µM of EGCG were selected for use in the subsequent experiments.
EGCG attenuates LPS-induced
inflammation in H9c2 cardiac cells
To investigate the effects of EGCG on the
inflammatory response in H9c2 cells, inflammatory cytokine
expressions were determined using ELISA. LPS (250 ng/ml)
significantly increased the TNF-α (Fig.
3A; P<0.001), ICAM-1 (Fig.
3B; P<0.001) and MMP-2 (Fig.
3C; P<0.001) protein levels in the medium supernatant
compared with the control, according to ELISA, whereas EGCG
suppressed the release of these cytokines in LPS-treated cells in a
dose-dependent manner (Table I). The
levels of TNF-α and MMP-2 were significantly alleviated after EGCG
intervention (≥12.5 µmol/l). Meanwhile, treatment with EGCG (≥6.25
µmol/l) significantly reduced the expression of ICAM-1 induced by
LPS.
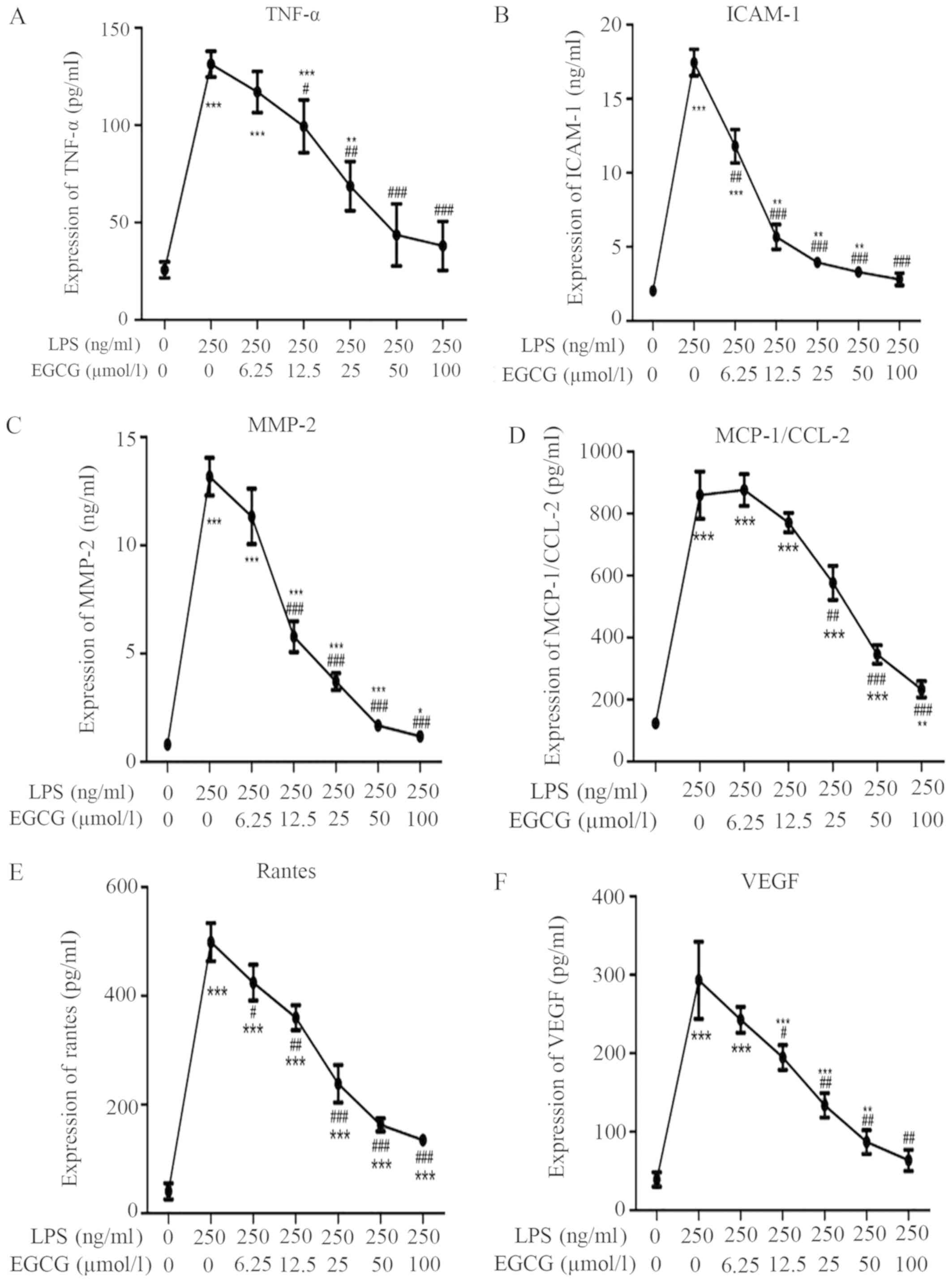 | Figure 3EGCG attenuates LPS-induced cytokine
release in H9c2 cells. (A) TNF-α content in the culture medium of
cultured H9c2 cells. (B) ICAM-1 content in the culture medium of
cultured H9c2 cells. (C) MMP-2 content in the culture medium of
cultured H9c2 cells. Effects of LPS and EGCG in H9c2 cells and the
supernatant levels of (D) MCP-1 and (E) Rantes of H9c2 incubated
with different concentrations of EGCG for 24 h were determined by
ELISA. (F) EGCG inhibited LPS-induced upregulation of VEGF. Data
are presented as the mean ± SEM. n=3 in each group.
*P<0.05, **P<0.01,
***P<0.001 vs. control group; #P<0.05,
##P<0.01, ###P<0.001 vs. LPS group.
EGCG, epigallocatechin-3-gallate; TNF-α, tumor necrosis factor-α;
ICAM-1, intercellular adhesion molecule-1; MMP-2, matrix
metalloproteinase 2; LPS, lipopolysaccharide; MCP-1, monocyte
chemotactic protein 1; VEGF, vascular endothelial growth
factor. |
 | Table IEffects of LPS and EGCG in H9c2 cells
and the supernatant levels of inflammatory mediators of H9c2
incubated with different concentrations of EGCG for 24 h were
determined by ELISA. |
Table I
Effects of LPS and EGCG in H9c2 cells
and the supernatant levels of inflammatory mediators of H9c2
incubated with different concentrations of EGCG for 24 h were
determined by ELISA.
| Groups Inflammatory
mediators | Control | 250 ng/ml LPS | 250 ng/ml LPS +
6.25 µmol/l EGCG | 250 ng/ml LPS +
12.5 µmol/l EGCG | 250 ng/ml LPS + 25
µmol/l EGCG | 250 ng/ml LPS + 50
µmol/l EGCG | 250 ng/ml LPS + 100
µmol/l EGCG |
|---|
| TNF-α, pg/ml | 25.67±4.16 |
131.33±6.66c |
117±10.54c |
99.33±13.58c,d |
68.67±12.66b,e |
43.67±15.95f |
38±12.53f |
| ICAM-1, ng/ml | 2.04±0.30 |
17.45±0.89c |
11.81±1.13c,e |
5.68±0.83b,f |
3.96±0.36b,f |
3.31±0.25b,f |
2.81±0.41f |
| MMP-2, ng/ml | 0.81±0.10 |
13.17±0.87c |
11.33±1.28c |
5.78±0.39c,f |
3.71±0.39c,f |
1.68±0.12c,f |
1.18±0.19a,f |
| MCP-1, pg/ml | 123.67±16.56 |
859.33±76.20c |
876±51.03c |
770.67±31.26c |
576.33±55.10c,e |
345.33±30.24a,f |
233.33±26.08b,f |
| Rantes, ng/ml | 40.67±14.74 |
498.67±35.16c |
424.33±33.01c |
359.67±22.89c,e |
238.33±34.65c,f |
162.67±12.10c,f |
134.67±6.51c,f |
| VEGF, pg/ml | 39.33±9.07 |
293±49.11c |
242.67±16.5c |
194.67±15.82c,d |
133.67±15.57c,e |
87±15.13b,e |
63.67±13.58e |
EGCG inhibits LPS-induced chemokine
expression related to inflammation
To further analyze the cardioprotective role of
EGCG, the concentrations of chemokines MCP-1 and Rantes in the H9c2
cell medium supernatant were analyzed after LPS stimulation. The
present data demonstrated that LPS induced a significant
upregulation of the expression of MCP-1 (Fig. 3D; P<0.001) and Rantes (Fig. 3E; P<0.001) compared with the
control group. EGCG treatment significantly attenuated the
LPS-induced increased MCP-1 and Rantes in a dose-dependent manner
when the concentrations of EGCG were ≥25 or ≥6.25 µmol/l,
respectively (Fig. 3D and E; Table
I).
Inhibitory effect of EGCG on
LPS-induced upregulation of VEGF in H9c2 cells
To evaluate the inhibitory effect of EGCG on the
LPS-induced upregulation of VEGF, the levels of VEGF in the
supernatant of H9c2 cells were determined by ELISA. Cultured H9c2
cells were treated without or with LPS (250 ng/ml), or LPS (250
ng/ml) + EGCG (6.25, 12.5, 25, 50 and 100 µmol/l) for 24 h. EGCG
(≥12.5 µmol/l) significantly diminished the LPS-induced upregulation
of VEGF (Fig. 3F).
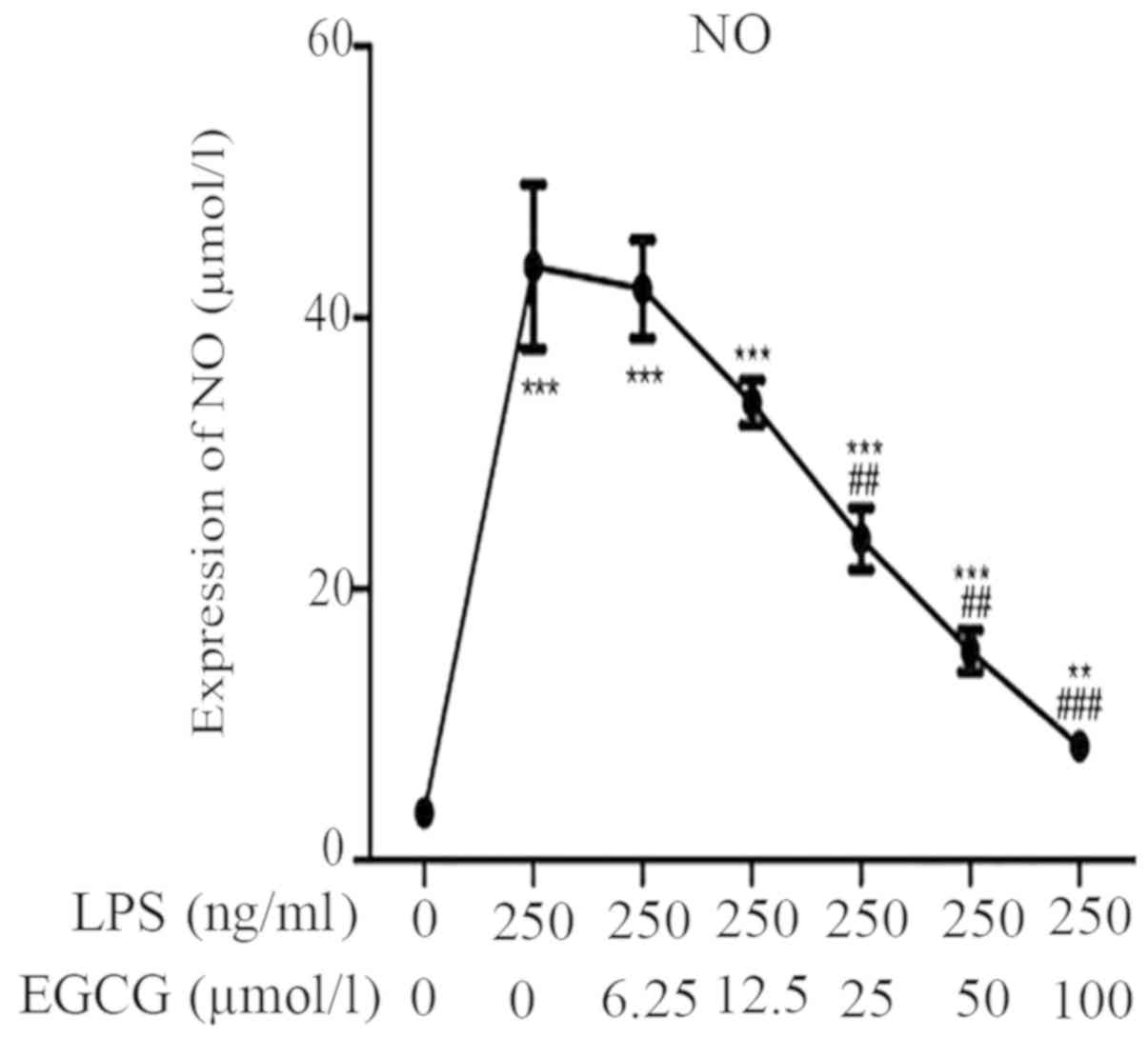
EGCG suppresses NO expression in
LPS-induced inflammation of H9c2 cardiomyocytes
The effect of EGCG on the NO content in the culture
medium was additionally determined. LPS significantly increased the
NO content in the culture medium (Fig.
4; Table II; P<0.001).
However, EGCG (≥25 µmol/l) significantly counteracted the induction
of NO release into the culture medium (Fig. 4).
| Table IIEGCG suppresses LPS-induced
upregulation of NO release. |
Table II
EGCG suppresses LPS-induced
upregulation of NO release.
| Group | NO, µmol/l |
|---|
| Control | 3.52±0.45 |
| 250 ng/ml LPS |
43.76±6.08a |
| 250 ng/ml LPS +
6.25 µmol/l EGCG |
42.11±3.62a |
| 250 ng/ml LPS +
12.5 µmol/l EGCG |
33.73±1.65a |
| 250 ng/ml LPS + 25
µmol/l EGCG |
23.70±2.29a,b |
| 250 ng/ml LPS + 50
µmol/l EGCG |
15.42±1.53a,b |
| 250 ng/ml LPS + 100
µmol/l EGCG |
8.42±1.13a,c |
Potential mechanisms of EGCG
inhibition of LPS-induced inflammatory responses in H9c2 cells
To further elucidate the mechanism underlying the
anti-inflammatory effects of EGCG on LPS-induced H9c2 cells,
western blot analysis was used to detect the potential pathways.
LPS (250 ng/ml) was identified to significantly increase p-Akt,
p-ERK, p-NF-κB p65 and p-p38 compared with the control (Fig. 5). Conversely, treatment with EGCG
(≥12.5 µmol/l) significantly suppressed the LPS-induced
inflammatory protein activation as demonstrated by the
downregulated phosphorylation of Akt (Fig. 5B), NF-κB p65 (Fig. 5D) and p38 (Fig. 5E) compared with the LPS group. The
induction of p-ERK expression by LPS was significantly reduced by
≥50 µmol/l EGCG (Fig. 5C).
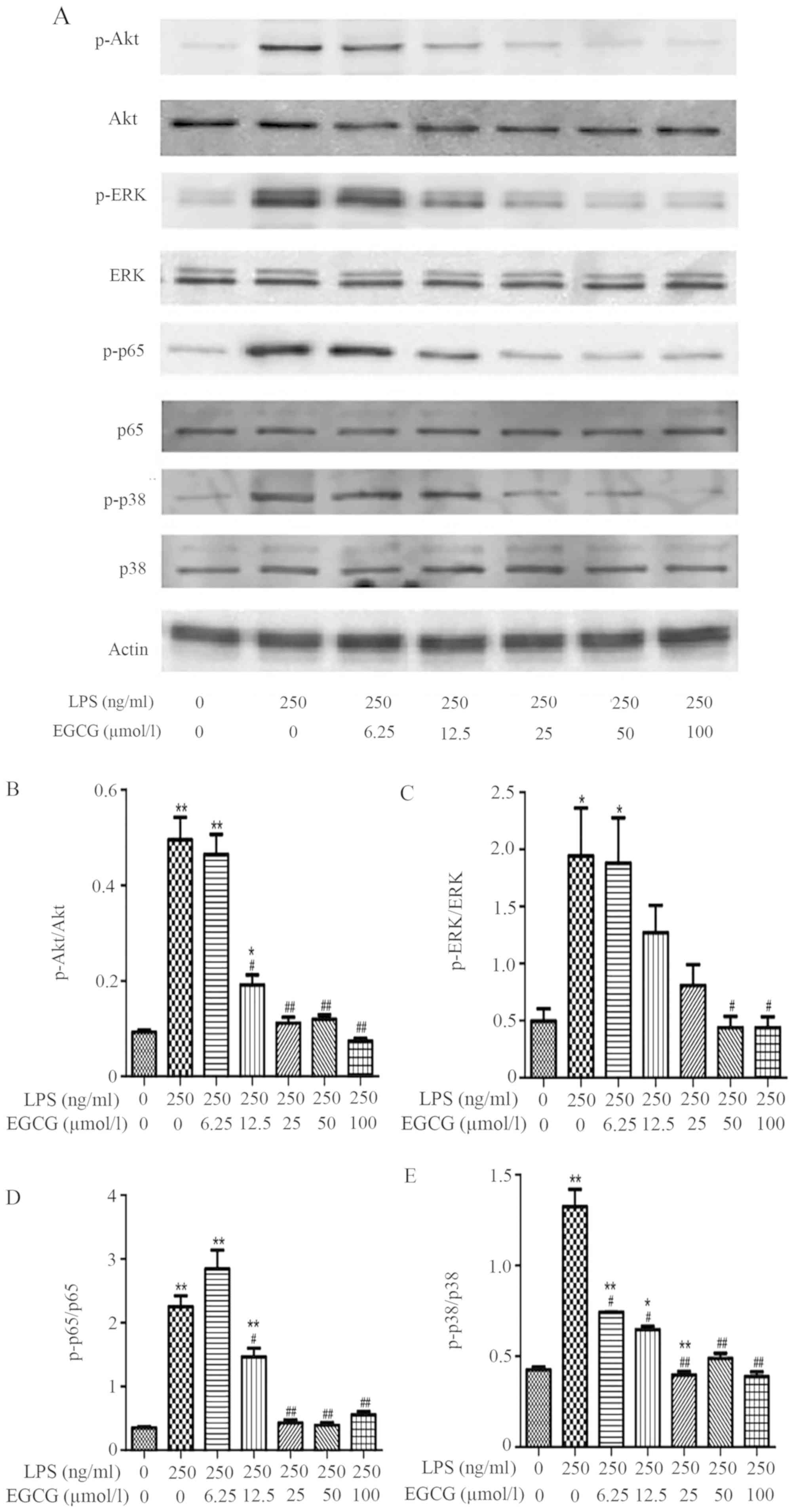 | Figure 5EGCG suppresses LPS-induced marked
inflammatory protein activation. Cultured H9c2 cells were treated
with control or LPS (250 ng/ml) or LPS + EGCG (0, 6.25, 12.5, 25,
50 and 100 µmol/l) for 24 h. After immunoblotting, the
phosphorylation or the total levels of Akt, ERK, p38, nuclear
factor-κB p65 were identified through their phosphor-specific or
non-phosphor-specific antibodies. (A) The protein expressions of
phosphorylated/total Akt, ERK, p38, nuclear factor-κB and p65 are
presented. The expressions of (B) p-Akt/Akt, (C) p-ERK/ERK, (D)
p-p65/p65 and (E) of p-p38/p38 are demonstrated. Data are presented
as the mean ± SEM. n=3 in each group. *P<0.05,
**P<0.01 vs. control group; #P<0.05,
##P<0.01 vs. LPS group. EGCG,
epigallocatechin-3-gallate; LPS, lipopolysaccharide; p,
phosphorylated. |
Discussion
The present study provided evidence that H9c2 cells
with LPS-induced inflammation, exhibited enhanced inflammatory
mediators, such as VEGF, Rantes, MCP-1, ICAM-1, MMP-2, TNF-α and
NO. However, the upregulation of the inflammatory mediators were
attenuated by EGCG treatment. Additionally, EGCG suppressed the
inflammatory signal pathway by downregulating p-NF-κB p65, p-Akt,
p-ERK and p-p38.
EGCG is the main and most significantly bioactive
polyphenol found in solid green tea extract, accounting for ~65% of
the catechin content; 250 mg EGCG is present in a brewed cup of
green tea (22). Several previous
studies showed that EGCG has important anti-atherogenic and
anti-inflammatory properties (23,24). The
anti-inflammatory effects of EGCG have been demonstrated in
numerous previous studies related to the pathological conditions
where inflammation is a core-driving factor (23,24). For
example, previous studies identified that EGCG was effective in
preventing IL-8 production in airway epithelial cells through
stimulation of IL-1β, restraining the development of respiratory
inflammation (25,26). Moreover, EGCG treatment ameliorated
cigarette smoke (CS)-induced airway inflammation and mucus secretion
in a CS-exposed rat model (27).
EGCG was also demonstrated to reduce cardiac apoptosis by
decreasing the inflammatory response (28) and attenuating the serum level of
cardiac function biomarker enzymes in rats (29). However, EGCG possesses several
limitations, such as poor stability and low bioavailability, which
are associated with the concentration of EGCG. High doses of EGCG
achieve a positive result of antioxidant and pro-oxidative
properties. However, a series of toxic side effects are induced by
high doses of EGCG (30). Previous
clinical studies have identified that the side effects of EGCG
include nausea, insomnia and hepatotoxicity (31,32).
When the present study was conducted, to the best of the authors'
knowledge there were no previous studies in the literature to check
the optimal EGCG concentration. Specific concentrations ranging
from 50-400 µM were adopted according to the cell viability
experiment in different literatures (33,34).
Therefore, in the present study the safest maximum concentration
(100 µM) was used in the present experiments. Recent studies on the
optimum concentrations have since been conducted, the cell
viability and Actin-Tracker Green technique in these recent studies
demonstrated that the optimal range of concentration for the
protective effects of EGCG was 50-100 µM (35,36).
H9c2 is a traditional cell line used to study
myocardial disease, which was preserved in the laboratory of
Hangzhou Red Cross Hospital/Hospital of Integrated Traditional
Chinese and Western Medicine. Moreover, animal cells are also one
of the most important ways to establish disease models, so that
research is not limited to using human cell lines. Many previous
studies on the effects of LPS on cardiomyocytes also used the H9c2
cell line (37-39).
Additionally, subsequent validation experiments were performed in
rats in addition to H9c2 cell lines. Therefore, the present study
selected the H9c2 cell line for the present investigations.
NO is generated from all cell types composing the
myocardium and regulates cardiac function, including coronary
vessel tone, proliferative and inflammatory properties (40). Under severe inflammatory conditions,
excessive NO production causes decreased vascular tonus and
consequently hypotension, which is a characteristic of sepsis
(19).
LPS, released from the surface of the cell membrane
of Gram-negative bacteria, contributes to an inflammatory response.
The cellular response to LPS includes the production of reactive
oxygen species and other mediators, such as NO and pro-inflammatory
cytokines (41). Oxidative stress, a
condition caused by excessive production of free radicals, plays an
important role in the progression of an inflammatory condition
(42). Previous clinical studies
demonstrated that the levels of TNF-α and serum concentration of
soluble tumor necrosis factor receptors and IL are increased in
patients with chronic HF (43-45).
Indeed, concentrations of TNF-α are associated with the stimulation
of NO synthase, contributing to reduced cardiac contractility in HF
(46). As for NO, three NO synthases
support various involvements of NO in cardiac physiology. Induced
excessive NO from inflammatory cells and LPS-stimulated
cardiomyocytes themselves, may lead to profound cellular
disturbances resulting in HF (47).
In the present study, with LPS-induced inflammation, H9c2 exhibited
upregulated expression of inflammatory factors and oxidative stress
molecules, such as Rantes, MCP-1, ICAM-1, MMP-2, TNF-α and NO.
Moreover, the effects of LPS (250 µg/ml) on cell viability were
examined and it was identified that LPS (250 µg/ml) had no effect
on the cell viability rate or cytotoxicity in H9c2 cells compared
with the control group (101.52±9.0% vs. 100%; P>0.05; data not
shown) and the chosen concentration of LPS (250 µg/ml) was lower
the concentration used in literature (48,49). The
pro-inflammatory cytokines play an important role in the inhibition
of cardiac function and the progression from cardiac injury to
failure (50). Treatment with EGCG
after LPS-stimulated inflammation significantly decreased the
levels of pro-inflammatory cytokines and adhesion molecules,
suggesting its anti-inflammatory potential.
To demonstrate how EGCG plays a role in the
inflammation process, the effects on the inflammatory response of
H9c2 cells stimulated by LPS were investigated. H9c2 cells
stimulated by LPS demonstrated an increased activation of p-Akt.
However, EGCG treatment inhibited the expression of p-Akt. PI3K/Akt
signaling is an important pathway involved in controlling
cardiomyocyte function and survival (51). One of the downstream effectors of Akt
in the PI3K/Akt pathway is endothelial NO synthase, which after
phosphorylation, leads to the production of NO (52). Excessive NO delivery from
inflammatory cells and cardiomyocytes may influence cellular
abnormal and cardiac contractility (40). The present results demonstrated that
EGCG significantly counteracted the induction of NO release into the
culture medium. Furthermore, the activation of p-p38, p-NF-κB p65
and p-ERK was increased by stimulation of LPS in H9c2 cells. EGCG
suppressed the activation of these phosphorylated proteins. NF-κB,
p38 and ERK are the most important factors playing a vital role in
mediating inflammatory responses to a variety of signals, including
inflammatory cytokines (53).
Therefore, these associated pathways may comprise important
molecular mechanisms responsible for cardiomyocyte inflammation
induced by LPS, and EGCG ameliorates LPS-induced inflammation in
H9c2 cells through the PI3K/Akt and p38 signaling pathways. A
potential mechanism is that EGCG affects the kinase upstream, and
then p38 and ERK regulate the downstream target NF-κB. Furthermore,
future studies should broaden the scope to determine the crosstalk
between the ERK and p38 pathways in H9c2 cells after EGCG treatment
by inhibiting ERK and p38.
In the present study, H9c2 cells were incubated
without or with EGCG at different concentrations for 48 h. The MTS
assay showed no effect on the cell viability with EGCG at
concentrations <200 µM. Although, further investigations will be
necessary to further study the effects of EGCG on cell viability in
H9c2 cells at multiple time points, such as 24, 48 and 96 h. The
present results should also be verified in human cardiomyocytes in
future studies.
The present findings provided evidence for the
inhibiting effects of EGCG on LPS-stimulated inflammation in H9c2
cells. These results suggested the therapeutic potential of EGCG in
cardiac inflammation.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Science Foundation for Youth of China (grant no. 30800533) and the
Zhejiang Provincial Natural Science Foundation of China (gran no.
LY14H070002).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL, ZS, HY, ST, XL and FW designed the present
study, analyzed the data and wrote and revised the manuscript.
Moreover, FW gave final approval of the version to be published.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Coggins M and Rosenzweig A: The fire
within: Cardiac inflammatory signaling in health and disease. Circ
Res. 110:116–25. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Marchant DJ, Boyd JH, Lin DC, Granville
DJ, Garmaroudi FS and McManus BM: Inflammation in myocardial
diseases. Circ Res. 110:126–144. 2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Płóciennikowska A, Hromada-Judycka A,
Borzęcka K and Kwiatkowska K: Co-operation of TLR4 and raft
proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life
Sci. 72:557–581. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wan QQ, Wu D and Ye QF: The expression
profiles of circRNAs in lung tissues from rats with
lipopolysaccharide-induced acute respiratory distress syndrome: A
microarray study. Biochem Biophys Res Commun. 493:684–689.
2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhang J, Yang S, Chen F, Li H and Chen B:
Ginkgetin aglycone ameliorates LPS-induced acute kidney injury by
activating SIRT1 via inhibiting the NF-κB signaling pathway. Cell
Biosci. 7(44)2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Frazier WJ, Xue J, Luce WA and Liu Y: MAPK
signaling drives inflammation in LPS-stimulated cardiomyocytes: The
route of crosstalk to G-protein-coupled receptors. PLoS One.
7(e50071)2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Goldberg RB: Cytokine and cytokine-like
inflammation markers, endothelial dysfunction, and imbalanced
coagulation in development of diabetes and its complications. J
Clin Endocrinol Metab. 94:3171–3182. 2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Cocco G, Jerie P, Amiet P and Pandolfi S:
Inflammation in heart failure: Known knowns and unknown unknowns.
Expert Opin Pharmacother. 18:1225–1233. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tschöpe C, Walther T, Escher F, Spillmann
F, Du J, Altmann C, Schimke I, Bader M, Sanchez-Ferrer CF,
Schultheiss HP and Noutsias M: Transgenic activation of the
kallikrein-kinin system inhibits intramyocardial inflammation,
endothelial dysfunction and oxidative stress in experimental
diabetic cardiomyopathy. FASEB J. 19:2057–2059. 2005.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ma XL, Kumar S, Gao F, Louden CS, Lopez
BL, Christopher TA, Wang C, Lee JC, Feuerstein GZ and Yue TL:
Inhibition of p38 mitogen-activated protein kinase decreases
cardiomyocyte apoptosis and improves cardiac function after
myocardial ischemia and reperfusion. Circulation. 99:1685–1691.
1999.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sun TL, Liu Z, Qi ZJ, Huang YP, Gao XQ and
Zhang YY: (-)-Epigallocatechin-3-gallate (EGCG) attenuates
arsenic-induced cardiotoxicity in rats. Food Chem Toxicol.
93:102–110. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yang CS, Lambert JD, Ju J, Lu G and Sang
S: Tea and cancer prevention: Molecular mechanisms and human
relevance. Toxicol Appl Pharmacol. 224:265–273. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Khan N, Afaq F, Saleem M, Ahmad N and
Mukhtar H: Targeting multiple signaling pathways by green tea
polyphenol (-)-epigallocatechin-3-gallate. Cancer Res.
66:2500–2505. 2006.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kuriyama S, Shimazu T, Ohmori K, Kikuchi
N, Nakaya N, Nishino Y, Tsubono Y and Tsuji I: Green tea
consumption and mortality due to cardiovascular disease, cancer,
and all causes in Japan: The Ohsaki study. JAMA. 296:1255–1265.
2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tseng CL, Hung YJ, Chen ZY, Fang HW and
Chen KH: Synergistic effect of artificial tears containing
epigallocatechin gallate and hyaluronic acid for the treatment of
rabbits with dry eye syndrome. PLoS One.
11(e0157982)2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hammad FT and Lubbad L: The effect of
epigallocatechin-3-gallate on the renal dysfunction in the
obstructed kidney in the rat. Int J Physiol Pathophysiol Pharmacol.
9:119–126. 2017.PubMed/NCBI
|
|
17
|
Bliksoen M, Kaljusto ML, Vaage J and
Stensløkken KO: Effects of hydrogen sulphide on
ischaemia-reperfusion injury and ischaemic preconditioning in the
isolated, perfused rat heart. Eur J Cardiothorac Surg. 34:344–349.
2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhang W, Suo M, Yu G and Zhang M:
Antinociceptive and anti-inflammatory effects of cryptotanshinone
through PI3K/Akt signaling pathway in a rat model of neuropathic
pain. Chem Biol Interact. 305:127–133. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Franceschelli S, Pesce M, Ferrone A, Gatta
DM, Patruno A, Lutiis MA, Quiles JL, Grilli A, Felaco M and
Speranza L: Biological effect of licochalcone C on the regulation
of PI3K/Akt/eNOS and NF-kappaB/iNOS/NO signaling pathways in H9c2
cells in response to LPS stimulation. Int J Mol Sci. 18:
pii(E690)2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li X, Jiang L, Lin S, He Y, Shen G, Cai Z,
Ling M, Ni J, Zhang H and Zhang M: Inhibition of mTORC1 renders
cardiac protection against lipopolysaccharide. Int J Clin Exp
Pathol. 7:8432–8442. 2014.PubMed/NCBI
|
|
21
|
Sah JF, Balasubramanian S, Eckert RL and
Rorke EA: Epigallocatechin-3-gallate inhibits epidermal growth
factor receptor signaling pathway. Evidence for direct inhibition
of ERK1/2 and AKT kinases. J Biol Chem. 279:12755–12762.
2004.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yang CS, Wang X, Lu G and Picinich SC:
Cancer prevention by tea: Animal studies, molecular mechanisms and
human relevance. Nat Rev Cancer. 9:429–439. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Ou HC, Song TY, Yeh YC, Huang CY, Yang SF,
Chiu TH, Tsai KL, Chen KL, Wu YJ, Tsai CS, et al: EGCG protects
against oxidized LDL-induced endothelial dysfunction by inhibiting
LOX-1-mediated signaling. J Appl Physiol (1985). 108:1745–1756.
2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xu H, Lui WT, Chu CY, Ng PS, Wang CC and
Rogers MS: Anti-angiogenic effects of green tea catechin on an
experimental endometriosis mouse model. Hum Reprod. 24:608–618.
2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kim IB, Kim DY, Lee SJ, Sun MJ, Lee MS, Li
H, Cho JJ and Park CS: Inhibition of IL-8 production by green tea
polyphenols in human nasal fibroblasts and A549 epithelial cells.
Biol Pharm Bull. 29:1120–1125. 2006.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ge M, Xiao Y, Chen H, Luo F, Du G and Zeng
F: Multiple antiviral approaches of (-)-epigallocatechin-3-gallate
(EGCG) against porcine reproductive and respiratory syndrome virus
infection in vitro. Antiviral Res. 158:52–62. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liang Y, Liu KWK, Yeung SC, Li X, Ip MSM
and Mak JCW: (-)-Epigallocatechin-3-gallate reduces cigarette
smoke-induced airway neutrophilic inflammation and mucin
hypersecretion in rats. Front Pharmacol. 8(618)2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Saeed NM, El-Naga RN, El-Bakly WM,
Abdel-Rahman HM, Salah ElDin RA and El-Demerdash E:
Epigallocatechin-3-gallate pretreatment attenuates
doxorubicin-induced cardiotoxicity in rats: A mechanistic study.
Biochem Pharmacol. 95:145–155. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Othman AI, Elkomy MM, El-Missiry MA and
Dardor M: Epigallocatechin-3-gallate prevents cardiac apoptosis by
modulating the intrinsic apoptotic pathway in isoproterenol-induced
myocardial infarction. Eur J Pharmacol. 794:27–36. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Peter B, Bosze S and Horvath R:
Biophysical characteristics of proteins and living cells exposed to
the green tea polyphenol epigallocatechin-3-gallate (EGCg): Review
of recent advances from molecular mechanisms to nanomedicine and
clinical trials. Eur Biophys J. 46:1–24. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Liu C, Li P, Qu Z, Xiong W, Liu A and
Zhang S: Advances in the antagonism of Epigallocatechin-3-gallate
in the treatment of digestive tract tumors. Molecules. 24:
pii(E1726)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhu W, Mei H, Jia L, Zhao H, Li X, Meng X,
Zhao X, Xing L and Yu J: Epigallocatechin-3-gallate mouthwash
protects mucosa from radiation-induced mucositis in head and neck
cancer patients: A prospective, non-randomised, phase 1 trial.
Invest New Drugs: Nov 7, 2019 (Epub ahead of print).
|
|
33
|
Seok JK, Lee JW, Kim YM and Boo YC:
Punicalagin and (-)-Epigallocatechin-3-Gallate rescue cell
viability and attenuate inflammatory responses of human epidermal
keratinocytes exposed to airborne particulate matter PM10. Skin
Pharmacol Physiol. 31:134–143. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Holczer M, Besze B, Zámbó V, Csala M,
Bánhegyi G and Kapuy O: Epigallocatechin-3-gallate (EGCG) promotes
autophagy-dependent survival via influencing the balance of
mTOR-AMPK pathways upon endoplasmic reticulum stress. Oxid Med Cell
Longev. 2018(6721530)2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ma Y, Hu Y, Wu J, Wen J, Li S, Zhang L,
Zhang J, Li Y and Li J: Epigallocatechin-3-gallate inhibits
angiotensin II-induced cardiomyocyte hypertrophy via regulating
Hippo signaling pathway in H9c2 rat cardiomyocytes. Acta Biochim
Biophys Sin (Shanghai). 51:422–430. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhang C, Liao P, Liang R, Zheng X and Jian
J: Epigallocatechin gallate prevents mitochondrial impairment and
cell apoptosis by regulating miR-30a/p53 axis. Phytomedicine.
61(152845)2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Magi S, Nasti AA, Gratteri S, Castaldo P,
Bompadre S, Amoroso S and Lariccia V: Gram-negative endotoxin
lipopolysaccharide induces cardiac hypertrophy: Detrimental role of
Na(+)-Ca(2+) exchanger. Eur J Pharmacol. 746:31–40. 2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Li Y, Liu X, Du A, Zhu X and Yu B: miR-203
accelerates apoptosis and inflammation induced by LPS via targeting
NFIL3 in cardiomyocytes. J Cell Biochem. 120:6605–6613.
2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Su Q, Yao J and Sheng C: Geniposide
Attenuates LPS-induced injury via Up-regulation of miR-145 in H9c2
cells. Inflammation. 41:1229–1237. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Massion PB, Feron O, Dessy C and Balligand
JL: Nitric oxide and cardiac function: Ten years after, and
continuing. Circ Res. 93:388–398. 2003.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Franceschelli S, Pesce M, Ferrone A,
Patruno A, Pasqualone L, Carlucci G, Ferrone V, Carlucci M, de
Lutiis MA, Grilli A, et al: A Novel biological role of α-mangostin
in modulating inflammatory response through the activation of
sirt-1 signaling pathway. J Cell Physiol. 231:2439–2451.
2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Franceschelli S, Pesce M, Ferrone A, De
Lutiis MA, Patruno A, Grilli A, Felaco M and Speranza L:
Astaxanthin treatment confers protection against oxidative stress
in U937 cells stimulated with lipopolysaccharide reducing
O2-production. PLoS One. 9(e88359)2014.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Hori M and Yamaguchi O: Is tumor necrosis
factor-alpha friend or foe for chronic heart failure? Circ Res.
113:492–494. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Khodadadi I, Vahedi MS, Abdi M, Daneshkhah
N, Rahbari R, Menbari S, Ahmadi D, Ahmadi A, Lahoorpour F,
Hakhamaneshi MS, et al: Evaluation of adenosine deaminase (ADA)
isoenzymes activity and tumor necrosis factor-α (TNFα)
concentration in chronic heart failure. EXCLI J. 13:58–66.
2014.PubMed/NCBI
|
|
45
|
Eskandari V, Amirzargar AA, Mahmoudi MJ,
Rahnemoon Z, Rahmani F, Sadati S, Rahmati Z, Gorzin F, Hedayat M
and Rezaei N: Gene expression and levels of IL-6 and TNFα in PBMCs
correlate with severity and functional class in patients with
chronic heart failure. Ir J Med Sci. 187:359–368. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Parish RC and Evans JD: Inflammation in
chronic heart failure. Ann Pharmacother. 42:1002–1016.
2008.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Everett BM, Cornel JH, Lainscak M, Anker
SD, Abbate A, Thuren T, Libby P, Glynn RJ and Ridker PM:
Anti-Inflammatory therapy with canakinumab for the prevention of
hospitalization for heart failure. Circulation. 139:1289–1299.
2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zhang H, Li H, Ge A, Guo E, Liu S and
Zhang L: Long non-coding RNA TUG1 inhibits apoptosis and
inflammatory response in LPS-treated H9c2 cells by down-regulation
of miR-29b. Biomed Pharmacother. 101:663–669. 2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Hao R and Su G, Sun X, Kong X, Zhu C and
Su G: Adiponectin attenuates lipopolysaccharide-induced cell injury
of H9c2 cells by regulating AMPK pathway. Acta Biochim Biophys Sin
(Shanghai). 51:168–177. 2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Gonzalez A, Ravassa S, Beaumont J, López B
and Díez J: New targets to treat the structural remodeling of the
myocardium. J Am Coll Cardiol. 58:1833–1843. 2011.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wu Y, Xia ZY, Meng QT, Zhu J, Lei S, Xu J
and Dou J: Shen-Fu injection preconditioning inhibits myocardial
ischemia-reperfusion injury in diabetic rats: Activation of eNOS
via the PI3K/Akt pathway. J Biomed Biotechnol.
2011(384627)2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Li Q, Shen L, Wang Z, Jiang HP and Liu LX:
Tanshinone IIA protects against myocardial ischemia reperfusion
injury by activating the PI3K/Akt/mTOR signaling pathway. Biomed
Pharmacother. 84:106–114. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Ibrahim AS, El-Remessy AB, Matragoon S,
Zhang W, Patel Y, Khan S, Al-Gayyar MM, El-Shishtawy MM and Liou
GI: Retinal microglial activation and inflammation induced by
amadori-glycated albumin in a rat model of diabetes. Diabetes.
60:1122–1133. 2011.PubMed/NCBI View Article : Google Scholar
|