Introduction
Acute myeloid leukemia (AML) is a biologically and
clinically heterogeneous disorder that is characterized by clonal
expansion of myeloid progenitors in the bone marrow and peripheral
blood (1). The incidence of AML
increases with age, and >60% of patients are at least 60 years
old at the time of diagnosis (2).
The 5-year overall survival (OS) rate of AML is 40% for patients
younger than 60, but only 10-20% for older patients (3,4). The
majority of patients who relapse after complete remission (CR) do
not survive for more than 5 years (5). Even though several studies have
reported improvements in the outcomes of younger AML patients over
the last 40 years (6-8),
the prognosis of patients aged ≥60 is poor and has remained
virtually unchanged (6).
Combination chemotherapy remains the primary choice
of AML treatment, and the achievement of CR is crucial for the
long-term survival of patients with AML. For more than four
decades, induction chemotherapy with cytarabine and an
anthracycline (‘7+3’ therapy) has been applied as the standard
chemotherapy regimen for patients newly diagnosed with classical
AML (9). However, the remission rate
of ‘7+3’ therapy is not satisfactory. Although CR is achieved in
50-75% of adult AML patients treated with the deoxycytidine analog
cytarabine (Ara-c) and an anthracycline antibiotic [including
daunorubicin or idarubicin, or anthracenedione mitoxantrone (MIT)],
only 20-30% of patients experience long-term disease-free survival
(DFS) (10).
MA therapy, which comprises MIT combined with Ara-c,
is now widely used for the treatment of elderly patients with AML
(11-13).
Ahmed et al (14) previously
reported the use of L-asparaginase (L-Asp) in combination with
other conventional chemotherapeutic drugs in the treatment of
elderly patients with refractory AML. The findings suggest that
L-Asp may prove effective in the treatment of AML, and that L-Asp
plus conventional chemotherapeutic drugs may offer beneficial
effects in elderly patients with refractory AML.
L-Asp has been widely used in the treatment of
pediatric leukemia since its antitumor activity was first
discovered, which significantly improves the survival rate of
children with leukemia (15).
Nonetheless, the antitumor mechanism of L-Asp remains unclear. It
is generally considered that leukemia cells lack sufficient
activity of asparagine synthetase (16). Whereas, L-Asp can potentially
decompose and deplete asparagine present in the blood surrounding
the leukemia cells, leading to the dysfunction of protein synthesis
and eventual cell death (17,18).
Autophagy is a biological process in which large
molecules and damaged organelles in the cytoplasm are degraded.
Despite being a recycling mechanism that assists cells in all
eviating nutrient stress, autophagy has been demonstrated to
regulate cell differentiation, cell death and the cell cycle
(19-21).
Microtubule-associated protein 1 light chain 3 [LC3;
a homolog of autophagy-related protein 8 (Atg8)] is a protein
normally localized to the autophagosome membrane (22). LC3 was originally identified as a
subunit of the neuronal microtubule-associated proteins (MAPs)
MAPlA and MAPlB, and its expression can regulate the microtubule
binding activity of MAPlA and MAPlB (23). There are three human LC3 isoforms
(LC3A, LC3B and LC3C) that undergo post-translational modifications
during autophagy (24). The
C-terminus of newly synthesized LC3 is normally cleaved by Atg4
protease to form the cytosolic soluble LC3-I (25). Following autophagosome formation,
LC3-I is modified by shearing and ubiquitination, before being
coupled with phosphatidylethanolamine on the surface of the
autophagosome membrane to form a membrane-bound LC3-II, located on
the inner and outer membranes of the autophagosome (24,26).
Unlike other Atg family of proteins located on autophagosome
membranes, LC3-II remains stable on autophagosome membranes until
it fuses with lysosomes, and is therefore usually used as a marker
for autophagy detection (27-29).
In a previous study, Song et al (30) reported asparaginase-induced
autophagic properties in chronic myeloid leukemia (CML) cell lines
K562 and KU812. Therefore, it was hypothesized that L-Asp may exert
antiproliferative effects on AML cells by inducing autophagy. In
the present study, the effects of L-Asp on cell proliferation,
apoptosis, morphological changes and the autophagic activity of AML
cell lines U937, HL-60 and KG-1a were investigated. In addition,
the effects of combining L-Asp priming with MA therapy were also
assessed. The aim of this study was to elucidate of the mechanism
of L-Asp efficacy and to investigate whether the combination of
L-Asp priming with MA therapy could be beneficial in AML
treatment.
Materials and methods
Cell culture
Human AML cell lines U937, HL-60, and KG-1a were
purchased from China Infrastructure of Cell Line Resources
(Institute of Basic Medical Sciences, Chinese Academy of Medical
Sciences). U937 cells were cultured in RPMI-1640 medium (SH30809,
Hyclone; GE Healthcare Life Sciences) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; 10099-141; Gibco; Thermo
Fisher Scientific, Inc.), whereas HL-60 and KG-1a cells were
maintained in IMDM medium (SH30228.01B; Hyclone; GE Healthcare Life
Sciences) supplemented with 20% FBS. All cells were cultured at
37˚C under 5% CO2.
Materials and reagents
Ara-c (trade name, Cytosar) was purchased from
Pfizer Inc. and diluted in the provided water for injection with
benzyl alcohol according to the manufacturer's protocol. MIT (trade
name, Militant) and L-Asp (derived from Escherichia coli)
were purchased from Sichuan Shenghe Pharmaceutical Co., Ltd. and
Changzhou Qianhong Bio-Pharma Co., Ltd., respectively, and were
diluted in sterile 0.9% sodium chloride solution when used.
Chloroquine (CQ; cat. no. 14774), primary antibodies
for western blotting: Rabbit LC-3A/B (D3U4C; cat. no. 12741) and
rabbit glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 14C10;
Rabbit; cat. no. 2118), were all purchased from Cell Signaling
Technology (Cell Signaling Technology, Inc.).
Treatment of AML cells U937, HL-60,
and KG-1a
The cells were cultured at a density of 5,000
cells/well in the 96-well plates (100 µl/well) at 37˚C. AML cell
lines U937, HL-60 and KG-1a were treated with Ara-c, MIT and/or
L-Asp following the attainment of steady-state exponential growth.
For all cells, when determining the half maximal inhibitory
concentrations (IC50), Ara-c and MIT were applied at
concentrations of 0-8 µg/ml for 24 h, and L-Asp was applied at
concentrations of 0-6.4 U/ml for 48 h. In subsequent experiments,
U937, HL-60 and KG-1a cells were treated with Ara-c + MIT or Ara-c
+ MIT + L-Asp at their respective IC50 for 24 and 48 h.
The IC50 values were calculated using GraphPad Prism
version 6.0 (GraphPad Software, Inc.).
Cell proliferation analysis
Following each drug treatment regimen, cell
viability was measured using Cell Counting kit-8 (CCK-8; CK04;
Dojindo Molecular Technologies, Inc.) according to the
manufacturer's protocols. Cell growth inhibition rates were
calculated as follows: Growth inhibition rate
(%)=[(Acontrol-Ablank of
control)-(Asample-Ablank of
sample)]/(Acontrol-Ablank of control).
A indicates the absorbance at 460 nm.
Flow cytometric analysis
AML cells (U937, HL-60 and KG-1a) cultured at a
density of 4x105 cells/well in 6 cm dishes were
separated into four different treatment groups: Control, Ara-c +
MIT, L-Asp and Ara-c + MIT + L-Asp. The control group was cultured
in media supplemented with 10% FBS for 48 h without drug treatment.
The Ara-c + MIT group was cultured in media with 10% FBS for 24 h
then treated with Ara-c and MIT for the next 24 h. The L-Asp group
was treated with L-Asp for 48 h. The Ara-c + MIT + L-Asp group was
treated with L-Asp alone for 24 h and then together with Ara-c and
MIT for the next 24 h. At the end of their respective treatment
regimen, cells were subsequently stained with Annexin V/FITC and
propidium iodide (PI) using an Annexin V-FITC Apoptosis Detection
kit (Sigma-Aldrich; Merck KGaA) according to the manufacturer's
protocol, and analyzed using the ACEA NovoCyte Flow Cytometer (ACEA
Biosciences; Agilent Technologies, Inc.). Each experiment was
repeated three times, and the mean values were applied to calculate
the rates of apoptosis induction for each treatment group. FlowJo
software (version 7.6.1, FlowJo LLC) was used for analysis.
Cell imaging
AML cells from the different treatment groups were
seeded into 10-cm dishes at the density of 1x106
cells/well. Cell morphology was assessed using Olympus BX63
(Olympus Corporation) under x100 and x400 magnification.
Protein extraction for autophagy
detection
In the present study, U937, HL-60 and KG-1a cells
were seeded into 6 cm dishes at a density of 4x105
cells/well, and divided into two groups: CQ+ and CQ-. The CQ+ group
was treated with 10 µM CQ for 30 min, followed by L-Asp for 0, 3,
6, 12, 24 and 48 h. The CQ-group was directly treated with L-Asp
for 0, 3, 6, 12, 24 and 48 h without CQ pre-treatment. For the CQ+
and CQ-groups, the concentration of L-Asp used was determined on
the basis of its IC50 value in each cell line. At each
time point, protein samples of the AML cells from each treatment
group were extracted for western blot analysis.
Western blot analysis
Confluent AML cells grown on 6 cm dishes were washed
with ice-cold PBS. Following centrifugation at 1,500 x g for 5 min
to discard the medium, 10 ml ice-cold PBS was added in and mixed
well with the cells in 10 ml centrifuge tubes. After a period of 5
min, centrifugation was performed at 1,500 x g to discard the PBS,
twice before subsequent lysisusing 2X SDS-PAGE sample buffer (100
mM 1M Tris-HCl pH 6.8, 2% 2-mercaptoethanol, 20% glycerol and 4%
SDS). Bicinchoninic acid assay (P0010S; Beyotime Institute of
Biotechnology) was used for protein quantification. The lysates
were subjected to separation by SDS-PAGE in 12% gels at 15 µg per
lane, before the separated proteins were transferred onto
Immobilon®-P PVDF (Millipore; EMD Millipore). Following
blocking in PBS containing 5% non-fat dried milk and 0.1% Tween-20
for 1 h at room temperature, the membranes were incubated overnight
at 4˚C with the LC3A/B antibody (14/16 kDa. #4108S; CST) using a
dilution of 1:1,000. On the same membranes, incubation with the
LC3A/B antibody was followed by 2 h incubation at room temperature
with primary GAPDH antibody (GAPDH; 14C10; Rabbit; cat. no. 2118)
using a dilution of 1:4,000. The membranes were subsequently
incubated for 2 h with goat anti-rabbit (cat. no. sc-2004; Santa
Cruz Biotechnology, Inc.) at a dilution of 1:5,000 at room
temperature. The protein bands were visualized using an ECL-Plus
kit (00161543; Thermo Fisher Scientific, Inc.) coupled with X-ray
radio graphs (038401501; Carestream Health, Inc.).
Statistical analysis
All statistical analysis was performed in GraphPad
Prism version 6.0 (GraphPad Software, Inc.). All data are presented
as the mean ± standard deviation (SD). One-way ANOVA followed by
Tukey's multiple comparison test was used to determine statistical
significance. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of L-Asp, Ara-c and MIT alone
on AML cell viability
To determine the growth inhibitory effect of L-Asp
on the AML cell lines U937, HL-60 and KG-1a, the cells were treated
with ascending concentrations for 48 h prior to analysis. L-Asp
significantly inhibited cell proliferation in all three AML cell
lines in a dose-dependent manner, and was even effective at 0.1
U/ml, which was the lowest concentration of tested (Fig. 1).The 48 h IC50 values of
L-Asp were calculated to be 0.106, 0.44 and 0.098 U/ml for U937,
HL-60 and KG-1a, respectively. Additionally, the
concentration-growth inhibition rate curves of Ara-c and MIT
treatments alone were produced for each of the three AML cell
lines. As presented in Fig. 2, Ara-c
inhibited cell proliferation in all three AML cell lines in a
dose-dependent manner, while the inhibition effect of MIT would
reach a threshold at certain concentrations (0.1-1 µg/ml). For
U937, HL-60 and KG-1a cells, the 24 h IC50 values of
Ara-c and MIT were calculated to be 0.809, 3.07, 0.002 µg/ml and
0.002, 0.021 and 0.068 µg/ml, respectively.
Effects of Ara-c + MIT and L-Asp +
Ara-c + MIT treatments as inhibitors of AML cell proliferation
AML cell lines U937, HL-60 and KG-1a were first
pretreated with L-Asp for 24 h and subsequently with Ara-c and MIT
additively for the next 24 h. Each of the three cell lines were
treated with drugs at concentrations corresponding to their
respective IC50 values (Table
I). The combined effects of L-Asp pretreatment and subsequent
Ara-c + MIT treatment were analyzed in terms of growth inhibition
rates, calculated using the aforementioned formula.
 | Table IIC50 values of Ara-c and
MIT for 24 h treatment and L-Asp for 48 h treatment in the AML cell
lines U937, HL-60 and KG-1a. |
Table I
IC50 values of Ara-c and
MIT for 24 h treatment and L-Asp for 48 h treatment in the AML cell
lines U937, HL-60 and KG-1a.
| Cell line | IC50
value |
|---|
| Ara-c (µg/ml) | MIT (µg/ml) | L-Asp (U/ml) |
|---|
| U937 | 0.809 | 0.002 | 0.106 |
| HL-60 | 3.07 | 0.021 | 0.44 |
| KG-1a | 0.002 | 0.068 | 0.098 |
Treatment with Ara-c + MIT and L-Asp alone inhibited
the proliferation of U937 cells by 50.67 and 67.34%, respectively,
while the inhibition of proliferation induced by treatment with
L-Asp and Ara-c + MIT was significantly greater at 88.84% (Fig. 3A). Anti-proliferative effects were
also observed in KG-1a cells, with the inhibition rates of 34.39,
45.70 and 67.04% for Ara-c + MIT, L-Asp alone and Ara-c + MIT +
L-Asp treatment, respectively (Fig.
3C). Finally, treatment with Ara-c + MIT or L-Asp alone exerted
suppressive effects on HL-60 cell proliferation, with inhibition
rates of 89.54 and 62.78% respectively; while combined Ara-c + MIT
+ L-Asp treatment exhibited the highest inhibitory effect in this
cell line, with an inhibition rate of 90.78% (Fig. 3B).
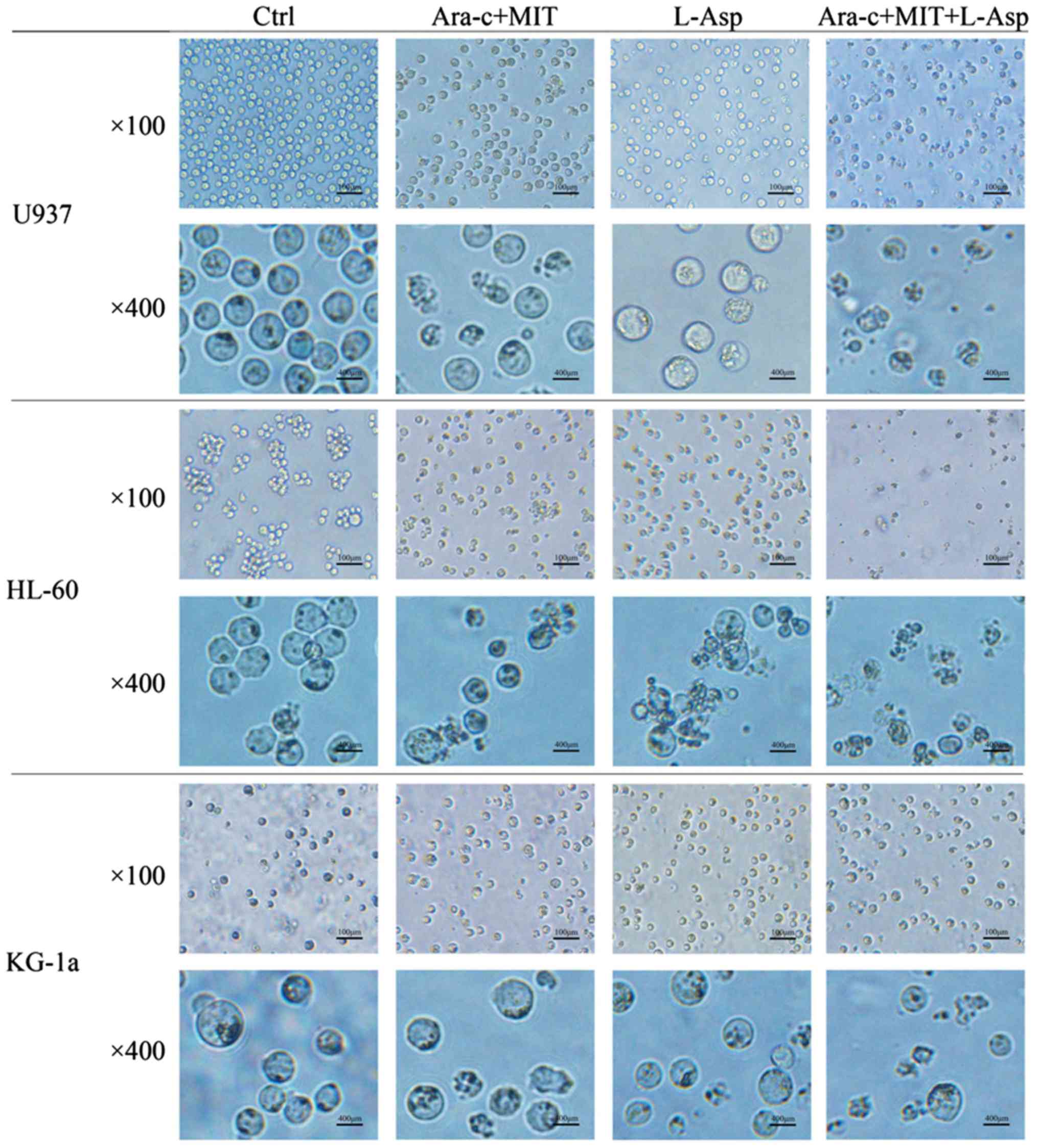
The morphological changes of AML cells from the
different treatment groups were examined using microscopy. In the
control group, U937, HL-60 and KG-1a cells all appeared circular
and bright with good refraction, whereas some of the cells that
were treated with Ara-c + MIT or L-Asp began to exhibit irregular
shapes and some granulation (Fig.
4). Notably, normally shaped living cells could barely be
observed in the medium in the Ara-c + MIT + L-Asp treatment group
(Fig. 4).
Effects of Ara-c + MIT and L-Asp +
Ara-c + MIT on the apoptosis of AML cells
Cell apoptosis was detected by flow cytometry using
Annexin V-FITC/PI dual staining. The results indicate that all
three treatment (Ara-c + MIT, L-Asp alone and Ara-c + MIT + L-Asp)
induced significant cell apoptosis in U937, HL-60 and KG-1a cells
compared with the control (Fig. 5).
However, cell apoptosis in the Ara-c + MIT + L-Asp treatment group
did not differ significantly from that of the Ara-c + MIT treatment
group or the L-Asp treatment group in U937 cells (Fig. 5A and B). Ara-c + MIT + L-Asp demonstrated
significantly increased cell apoptosis compared with Ara-c + MIT or
L-Asp alone in HL-60 cells (Fig. 5A
and C); whereas Ara-c + MIT + L-Asp
treatment appeared to have potentiated cell apoptosis compared with
Ara-c + MIT treatment, but no statistical significance was observed
between Ara-c + MIT + L-Asp and L-Asp alone in KG-1a cells
(Fig. 5A and D).
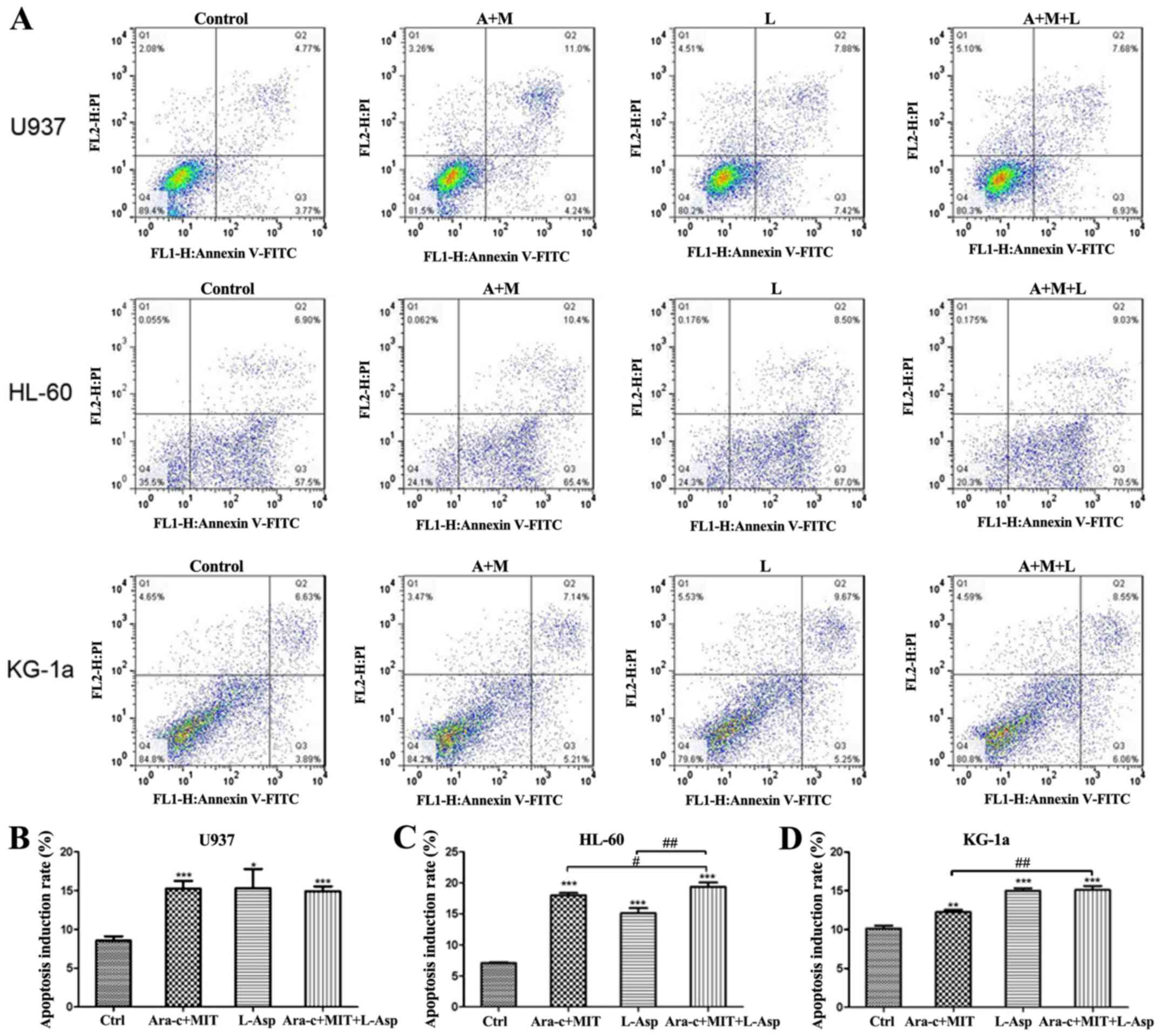 | Figure 5Comparison of cell apoptosis
induction rates in the Ara-c + MIT, L-Asp alone and Ara-c + MIT +
L-Asp treatment groups. (A) Flow cytometry dot blots show the
numbers of apoptotic cells as evaluated using Annexin V-FITC and PI
in U937, HL-60 and KG-1a cells. (B) Ara-c + MIT + L-Asp treatment
did not induce a difference in cell apoptosis compared with the
Ara-c + MIT or L-Asp alone groups in U937 cells. (C) Ara-c + MIT +
L-Asp treatment significantly increased apoptosis compared with
that in the Ara-c + MIT and L-Asp treatment groups in HL-60 cells.
(D) Ara-c + MIT + L-Asp significantly increased cell apoptosis
compared with that in the Ara-c + MIT group, but not with the L-Asp
group. Each experiment was conducted three times (n=3).
*P<0.05, **P<0.005,
***P<0.001 vs. Ctrl.
#P<0.01, ##P<0.001 as indicated. Ctrl,
control; Ara-c, cytarabine; MIT, mitoxantrone; L-Asp,
L-asparaginase; FITC, fluorescein isothiocyante; PI, propidium
iodide. |
Autophagy detection
To evaluate the effects of L-Asp on autophagy on the
U937, HL-60 and KG-1a cell lines, western blot analysis was used to
assess the ratio of autophagy-associated protein LC3A/B expression
levels in the cells (Fig. 6A). Each
cell line was divided into two groups, namely a CQ+ group that was
treated with 10 µM CQ for 3 h prior to protein extraction, and a
CQ-group that was not treated with CQ. In both treatment groups,
cells were treated with L-Asp at a concentration of 0.1 U/ml for 0,
3, 6, 12, 24 and 48 h. A time-dependent increase in LC3A/B-II/I
ratio following L-Asp treatment was only observed in U937 cells,
and not in HL-60 and KG-1a cells (Fig.
6B-D).
Discussion
AML is a malignant myeloproliferative disease that
is derived from hematopoietic stem cells (HSCs). It accounts for
~70% of acute leukemia cases and has an incidence of
2-4/100,000(31). Although
chemotherapy, targeted therapy, and HSC transplantation (32-35)
have all conferred improvements in CR and long-term DFS, the
majority of AML patients ultimately relapse and develop refractory
leukemia due to primary/secondary drug resistance, leading to
treatment failure (4,5). The etiology of AML is complex, and its
pathogenesis is not completely understood. In light of the high
mortality associated with AML and the lack of significant
improvements in prognosis over the last 30 years (36,37),
novel therapeutic approaches for AML are required.
In the present study, it was observed that L-Asp
treatment alone inhibited cell proliferation and induced apoptosis
in the AML cell lines U937, HL-60 and KG-1a. The effects of L-Asp +
Ara-c + MIT were compared those of L-Asp alone and Ara-c+ MIT.
Combining L-Asp with Ara-c + MIT in the treatment procedure
potentiated the inhibition of AML cell proliferation in each of the
three cell lines investigated, and resulted in greater cell
apoptosis in HL-60 and KG-1a cells. The present study investigated
the effects of L-Asp on AML cell lines, and the results support the
suggestion that the combination of L-Asp, MIT and Ara-c could be a
promising new therapeutic strategy for AML.
Autophagy is an evolutionary conserved process that
occurs throughout the eukaryotic phylogenetic tree and is essential
for cellular survival and development (38). Aside from the maintenance of
homeostasis, this catabolic process protects organisms from
conditions including neurodegenerative diseases, heart disease,
liver disease and aging (39).
It is generally recognized that autophagy can have
favorable and unfavorable outcomes in tumorigenesis and metastasis
(40). Previous studies demonstrated
that basal autophagy activation can modulate downstream signaling
pathways that are involved in maintaining adequate HSC
proliferation and differentiation (41). Impaired autophagy can result in the
transformation of HSCs into a preleukemic state, increasing the
likelihood of malignant leukemic transformation (42). Previous studies by Karvela et
al (42) and Rothe et al
(43) illustrated that autophagy may
serve a carcinogenic role in CML. Therefore, autophagy may serve
diverse functions in leukemia physiology.
L-Asp may serve a role autophagy in AML cells.
Several studies have implicated autophagic effects of L-Asp in a
number of malignancies including leukemia. Yu et al
(44) and Song et al
(45) proposed that L-Asp could
induce autophagy in ovarian cancer and CML, potentially
contributing to antitumor effects. Additionally, other studies have
demonstrated that suppressing autophagy may improve the
antineoplastic effect of L-Asp in glioblastoma, whereas activating
this process could overcome the L-Asp-induced immune suppression in
macrophages (39); suggesting that
L-Asp treatment may induce autophagy by promoting apoptosis, and
inhibiting cell growth in AML cells.
LC3-II content or the LC3-II/LC3-I ratio is
positively correlated with the number of autophagosomes and the
autophagic activity of cells to some extent (46-48).
In the present study, the LC3-II/I ratio was used to evaluate the
autophagic activity of cells.
Since autophagy is a dynamic process, the static
level of LC-3II/I alone is not sufficient to accurately measure
changes in intracellular autophagic flux. Chloroquine (CQ), which
is a lysosome inhibitor that inhibits autophagosome-lysosome fusion
(31), is commonly used for studying
autophagy and autophagy flux. To this end, changes in autophagy, as
indicated by the LC3 A/B expression ratio, were evaluated in AML
cells treated with L-Asp with and without CQ. However, inconsistent
and in conclusive autophagic effects were observed in the three AML
cell lines following L-Asp treatment, indicating that the
L-Asp-induced effects on AML cells may be attributed to alternative
mechanisms. Also, as this time-dependent response occurred in CQ-
and CQ+ groups, the increase in LC3A/B II/I ratio presented in
Fig. 6B-D may be due to factors
other than autophagy. Therefore, whether L-Asp treatment produced
any notable changes in autophagy in the three AML cell lines
warrants further investigation. And additional experimental methods
should be performed to unravel the signaling pathways mediating the
anti-proliferative and pro-apoptotic effects of L-Asp on AML
cells.
In conclusion, data from the present study
demonstrated that L-Asp significantly suppressed cell growth while
increasing apoptosis in the AML cell lines U937, HL-60 and KG-1a.
When L-Asp was combined with Ara-c and MIT, these physiological
effects were potentiated further. These results support the
suggestion that a combination of L-Asp and MA may be useful as an
AML therapy.
Acknowledgements
The authors would like to thank the China-America
Institute of Neuroscience, Beijing Luhe Hospital, Capital Medical
University (Beijing, China) for their valuable technical assistance
with the experiments.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TC performed most of the experiments in the present
study and analyzed the obtained data. JZ and HuZ interpreted the
data. YuZ, YoZ and XZ helped TC to perform the CCK8 experiments.
HeZ was a major contributor to conceiving of the study and
participated in writing the manuscript.
Ethics approval and consent to
participate
The current study was performed in accordance with
the Declaration of Helsinki and was approved by the Ethics
Committee of Beijing Luhe Hospital, Capital Medical University
(Beijing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Saultz JN and Garzon R: Acute myeloid,
leukemia: A concise review. J Clin Med. 5(pii: E33)2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Cartwright RA and Staines A: Acute
leukaemias. Baillieres Clin Haematol. 5:1–26. 1992.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dohner H, Estey EH, Amadori S, Appelbaum
FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson
RA, et al: Diagnosis and management of acute myeloid leukemia in
adults: Recommendations from an international expert panel, on
behalf of the European Leukemia Net. Blood. 115:453–474.
2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Dohner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152.
2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Stein EM and Tallman MS: Emerging
therapeutic drugs for AML. Blood. 127:71–78. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Xie Y, Davies SM, Xiang Y, Robison LL and
Ross JA: Trends in leukemia incidence and survival in the United
States (1973-1998). Cancer. 97:2229–2235. 2003.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Luke C, Nguyen AM, To B, Seshadri R,
Hughes T, Bardy P, Colbeck M, Buranyi-Trevarton D, McMellon M and
Roder D: Myeloid leukaemia treatment and survival-the South
Australian experience, 1977 to 2002. Asian Pac J Cancer Prev.
7:227–233. 2006.PubMed/NCBI
|
|
8
|
Norsworthy KJ, DeZern AE, Tsai HL, Hand
WA, Varadhan R, Gore SD, Gojo I, Pratz K, Carraway HE, Showel M, et
al: Timed sequential therapy for acute myelogenous leukemia:
Results of a retrospective study of 301 patients and review of the
literature. Leuk Res. 61:25–32. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Khaled S, Al-Malki M and Marcucci G: Acute
myeloid leukemia: Biologic, prognostic, and therapeutic insights.
Oncology (Williston Park). 30:318–329. 2016.PubMed/NCBI
|
|
10
|
Tallman MS, Gilliland DG and Rowe JM: Drug
therapy for acute myeloid leukemia. Blood. 106:1154–1163.
2005.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zeidner JF, Foster MC, Blackford AL,
Litzow MR, Morris LE, Strickland SA, Lancet JE, Bose P, Levy MY,
Tibes R, et al: Final results of a randomized multicenter phase II
study of alvocidib, cytarabine and mitoxantrone versus cytarabine
and daunorubicin (7 + 3) in newly diagnosed high-risk acute myeloid
leukemia (AML). Leuk Res. 72:92–95. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Saini NY, Cerny J, Furtado VF, Desmond A,
Zhou Z, Raffel G, Puthawala I, Bednarik J, Shanahan L, Miron PM, et
al: Elderly do benefit from induction chemotherapy: High dose
mitoxantrone-based (‘5 + 1’) induction chemotherapy regimen in
newly diagnosed acute myeloid leukemia. Am J Hematol. 94:209–215.
2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Pardee TS, Anderson RG, Pladna KM, Isom S,
Ghiraldeli LP, Miller LD, Chou JW, Jin G, Zhang W, Ellis LR, et al:
A Phase I study of CPI-613 in combination with high-dose cytarabine
and mitoxantrone for relapsed or refractory acute myeloid leukemia.
Clin Cancer Res. 24:2060–2073. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ahmed T, Holwerda S, Klepin HD, Isom S,
Ellis LR, Lyerly S, Manuel M, Dralle S, Berenzon D, Powell BL and
Pardee TS: High dose cytarabine, mitoxantrone and l-asparaginase
(HAMA) salvage for relapsed or refractory acute myeloid leukemia
(AML) in the elderly. Leuk Res. 39:945–949. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jia LT, Chen SY and Yang AG: Cancer gene
therapy targeting cellular apoptosis machinery. Cancer Treat Rev.
38:868–876. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dübbers A, Würthwein G, Müller HJ,
Schulze-Westhoff P, Winkelhorst M, Kurzknabe E, Lanvers C, Pieters
R, Kaspers GJ, Creutzig U, et al: Asparagine synthetase activity in
paediatric acute leukaemias: AML-M5 subtype shows lowest activity.
Br J Haematol. 109:427–429. 2000.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Baud V and Karin M: Signal transduction by
tumor necrosis factor and its relatives. Trends Cell Biol.
11:372–377. 2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Shen HM and Pervaiz S: TNF receptor
superfamily-induced cell death: Redox-dependent execution. FASEB J.
20:1589–1598. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Galluzzi L, Pietrocola F, Bravo-San Pedro
JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J,
Gewirtz DA, Karantza V, et al: Autophagy in malignant
transformation and cancer progression. EMBO J. 34:856–880.
2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Puissant A, Robert G and Auberger P:
Targeting autophagy to fight hematopoietic malignancies. Cell
Cycle. 9:3470–3478. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lin L and Baehrecke EH: Autophagy, cell
death, and cancer. Mol Cell Oncol. 2(e985913)2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tanida I, Sou YS, Minematsu-Ikeguchi N,
Ueno T and Kominami E: Atg8L/Apg8L is the fourth mammalian modifier
of mammalian Atg8 conjugation mediated by human Atg4B, Atg7 and
Atg3. FEBS J. 273:2553–2562. 2006.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Mann SS and Hammarback JA: Molecular
characterization of light chain 3. A microtubule binding subunit of
MAP1A and MAP1B. J Biol Chem. 269:11492–11497. 1994.PubMed/NCBI
|
|
24
|
Wu J, Dang Y, Su W, Liu C, Ma H, Shan Y,
Pei Y, Wan B, Guo J and Yu L: Molecular cloning and
characterization of rat LC3A and LC3B-two novel markers of
autophagosome. Biochem Biophys Res Commun. 339:437–442.
2006.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shpilka T, Weidberg H, Pietrokovski S and
Elazar Z: Atg8: An autophagy-related ubiquitin-like protein family.
Genome Biol. 12(226)2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
He H, Dang Y, Dai F, Guo Z, Wu J, She X,
Pei Y, Chen Y, Ling W, Wu C, et al: Post-translational
modifications of three members of the human MAP1LC3 family and
detection of a novel type of modification for MAP1LC3B. J Biol
Chem. 278:29278–29287. 2003.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Sou YS, Tanida I, Komatsu M, Ueno T and
Kominami E: Phosphatidylserine in addition to
phosphatidylethanolamine is an in vitro target of the mammalian
Atg8 modifiers, LC3, GABARAP, and GATE-16. J Biol Chem.
281:3017–3024. 2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Taguchi-Atarashi N, Hamasaki M, Matsunaga
K, Omori H, Ktistakis NT, Yoshimori T and Noda T: Modulation of
local PtdIns3P levels by the PI phosphatase MTMR3 regulates
constitutive autophagy. Traffic. 11:468–478. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Tanida I: Autophagy basics. Microbiol
Immunol. 55:1–11. 2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Song P, Ye L, Fan J, Li Y, Zeng X, Wang Z,
Wang S, Zhang G, Yang P, Cao Z and Ju D: Asparaginase induces
apoptosis and cytoprotective autophagy in chronic myeloid leukemia
cells. Oncotarget. 6:3861–3873. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yoshii SR and Mizushima N: Monitoring and
measuring autophagy. Int J Mol Sci. 18(pii: E1865)2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Gollner S, Oellerich T, Agrawal-Singh S,
Schenk T, Klein HU, Rohde C, Pabst C, Sauer T, Lerdrup M, Tavor S,
et al: Loss of the histone methyltransferase EZH2 induces
resistance to multiple drugs in acute myeloid leukemia. Nat Med.
23:69–78. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
33
|
Hourigan CS, Gale RP, Gormley NJ,
Ossenkoppele GJ and Walter RB: Measurable residual disease testing
in acute myeloid leukaemia. Leukemia. 31:1482–1490. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Xu B, Zhao Y, Wang X, Gong P and Ge W:
MZH29 is a novel potent inhibitor that overcomes drug resistance
FLT3 mutations in acute myeloid leukemia. Leukemia. 31:913–921.
2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Pollyea DA and Jordan CT: Therapeutic
targeting of acute myeloid leukemia stem cells. Blood.
129:1627–1635. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Estey EH: Treatment of acute myeloid
leukemia. Haematologica. 94:10–16. 2009.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Subramanian S and Kartha RV:
MicroRNA-mediated gene regulations in human sarcomas. Cell Mol Life
Sci. 69:3571–3585. 2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kabat AM, Pott J and Maloy KJ: The mucosal
immune system and its regulation by autophagy. Front Immunol.
7(240)2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ndoye A and Weeraratna AT: Autophagy-An
emerging target for melanoma therapy. F1000Res. 5(pii:
F1000)2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Li CJ, Liao WT, Wu MY and Chu PY: New
insights into the role of autophagy in tumor immune
microenvironment. Int J Mol Sci. 18(pii: E1566)2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhang WX, Zhou JC, Peng D, Hua F, Li K, Yu
JJ, Lv XX, Cui B, Liu SS, Yu JM, et al: Disrupting the TRIB3-SQSTM1
interaction reduces liver fibrosis by restoring autophagy and
suppressing exosome-mediated HSC activation. Autophagy. 1–15.
2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Karvela M, Baquero P, Kuntz EM,
Mukhopadhyay A, Mitchell R, Allan EK, Chan E, Kranc KR, Calabretta
B, Salomoni P, et al: ATG7 regulates energy metabolism,
differentiation and survival of Philadelphia-chromosome-positive
cells. Autophagy. 12:936–948. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Rothe K, Lin H, Lin KB, Leung A, Wang HM,
Malekesmaeili M, Brinkman RR, Forrest DL, Gorski SM and Jiang X:
The core autophagy protein ATG4B is a potential biomarker and
therapeutic target in CML stem/progenitor cells. Blood.
123:3622–3634. 2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Yu Y, Zhang X, Tian H, Zhang Z and Tian Y:
Knockdown of long non-coding RNA HOTAIR increases cisplatin
sensitivity in ovarian cancer by inhibiting cisplatin-induced
autophagy. J BUON. 23:1396–1401. 2018.PubMed/NCBI
|
|
45
|
Song X, Liu L, Chang M, Geng X, Wang X,
Wang W, Chen TC, Xie L and Song X: NEO212 induces mitochondrial
apoptosis and impairs autophagy flux in ovarian cancer. J Exp Clin
Cancer. 38(239)2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Gong C, Bauvy C, Tonelli G, Yue W,
Deloménie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V,
Tharinger H, et al: Beclin 1 and autophagy are required for the
tumorigenicity of breast cancer stem-like/progenitor cells.
Oncogene. 322261–2672. (2272e.1-11)2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Mitter SK, Song C, Qi X, Mao H, Rao H,
Akin D, Lewin A, Grant M, Dunn W Jr, Ding J, et al: Dysregulated
autophagy in the RPE is associated with increased susceptibility to
oxidative stress and AMD. Autophagy. 10:1989–2005. 2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lu Z, Chen C, Wu Z, Miao Y, Muhammad I,
Ding L, Tian E, Hu W, Ni H, Li R, et al: A dual role of P53 in
regulating colistin-induced autophagy in PC-12 cells. Front
Pharmacol. 8(768)2017.PubMed/NCBI View Article : Google Scholar
|